En este ejercicio clínico se presenta un caso que es discutido por un médico internista al que se le van proporcionando datos de la historia clínica en forma secuencial, y este analiza el cuadro a la luz de los nuevos elementos, de una manera análoga al proceso diagnóstico en la práctica real de la medicina
Un hombre de 48 años con diabetes mellitus tipo 2 de
larga evolución (nivel reciente de hemoglobina glicosilada, 6,5 %) y enfermedad
renal crónica (nivel de creatinina basal, 3,3 mg por decilitro; tasa de
filtración glomerular, 24 ml por minuto por 1,73 m 2 de superficie corporal) se
presentó a su médico de atención primaria con un historial de 3 meses de
entumecimiento, hormigueo y leve coloración violácea en las puntas de múltiples
dedos de manos y pies. Su examen físico mostró una sensación de tacto ligero
reducida en una distribución de guantes y medias; se palpaban los pulsos radial
y pedio. La vitamina B 12el nivel fue de 260 pg por mililitro (192 pmol por
litro; rango normal, 190 a 950 pg por mililitro [140 a 701 pmol por litro]). No
fumaba tabaco, no bebía alcohol ni consumía drogas ilícitas. Un mes después, se
desarrolló una herida no traumática en el pie izquierdo. El índice tobillo-brazo
(ITB) fue de 1,2 en ambos lados (rango normal, 0,91 a 1,3). Se inició el
cuidado de la herida por una presunta úlcera neuropática.
PONENTE
La parestesia y los déficits sensoriales son
característicos de una neuropatía de fibras pequeñas. La diabetes mellitus es
una causa frecuente de polineuropatía sensitiva distal. Sin embargo, la
afectación de las manos en combinación con síntomas en los pies que se limitan
a los dedos de los pies es inusual. Otras causas comunes de neuropatía
sensorial incluyen el consumo de alcohol, la deficiencia de vitaminas y el uso
de ciertos medicamentos; muchas neuropatías sensoriales no tienen explicación.
Un nivel de vitamina B 12 en suero bajo o normal debería impulsar la medición
del nivel de ácido metilmalónico para evaluar con mayor precisión la deficiencia
de vitamina B 12. Los pulsos periféricos palpables y el ITB de 1,2 sugieren que
la enfermedad macrovascular no es responsable de los síntomas en los dedos de
manos y pies. Sin embargo, las arterias altamente calcificadas (que son comunes
en el contexto de la diabetes y la enfermedad renal crónica) pueden no ser
comprimibles y, por lo tanto, pueden conducir a una elevación falsa del ITB a
pesar de la presencia de enfermedad arterial periférica. El índice dedo del
pie-brazo (IDB) puede ser una medida más precisa de la perfusión distal en el
contexto de estas enfermedades.
La coloración en los dedos de manos y pies podría
reflejar una interrupción en la microcirculación (p. ej., vasoespasmo o
crioglobulinemia). Las úlceras en los pies suelen surgir de una insuficiencia
arterial o venosa o de una lesión relacionada con la neuropatía. La enfermedad
renal crónica del paciente probablemente fue causada por su diabetes, pero se
justifica una evaluación adicional para confirmar que no es parte de un proceso
sistémico que también explicaría las ulceraciones y la neuropatía (p. ej.,
granulomatosis con poliangitis).
EVOLUCIÓN
Aproximadamente 1 mes después de la aparición de la
úlcera en el pie izquierdo, el paciente ingresó en un hospital con disnea
aguda. Su esposa notó que con frecuencia había estado somnoliento y menos
activo en general en las semanas anteriores. Refería exacerbación de lumbalgia
crónica, por lo que había estado tomando ibuprofeno intermitentemente durante
el mes anterior. Su otro historial médico incluía hiperlipidemia y obesidad
(que había sido tratada con bypass gástrico en Y de Roux 13 años antes). Estaba
tomando aspirina en dosis de 81 mg diarios, atorvastatina en dosis de 40 mg
diarios, gabapentina en dosis de 200 mg tres veces al día y un multivitamínico.
Su padre tenía diabetes mellitus tipo 2 y su madre demencia avanzada.
La temperatura oral del paciente era de 37,1°C, la
frecuencia cardíaca de 94 latidos por minuto, la presión arterial de 92/50 mm
Hg, la frecuencia respiratoria de 28 por minuto y la saturación de oxígeno de
93% mientras respiraba aire ambiente. El examen reveló edema con fóvea en ambos
pies y una escara que se había formado sobre la úlcera inicial en el mediopié
dorsal izquierdo que se extendía hasta las puntas de los primeros tres dedos
sin eritema ni induración circundantes.
El nivel de hemoglobina fue de 6,8 g por decilitro
(nivel inicial, 7,5); el recuento de glóbulos blancos y el recuento de
plaquetas eran normales. El nivel de potasio era de 6 mmol por litro y el nivel
de creatinina de 5,6 mg por decilitro (495 μmol por litro) (tasa de filtración
glomerular estimada, 12 ml por minuto). El nivel de bilirrubina total fue de
1,1 mg por decilitro (18,8 μmol por litro; rango normal, 0,3 a 1,2 mg por
decilitro [5,1 a 20,5 μmol por litro]), el nivel de lactato deshidrogenasa 196
U por litro (rango normal, 105 a 290) , y el nivel de haptoglobina 114 mg por
decilitro (rango normal, 50 a 200 mg por decilitro). El nivel de troponina T
fue de 0,03 ng por mililitro (rango normal, 0 a 0,04). No se obtuvo un frotis
de sangre. El análisis de orina mostró proteína 2+. Dos conjuntos secuenciales
de hemocultivos resultaron estériles. Un electrocardiograma mostró ritmo
sinusal. La ecografía renal descartó el diagnóstico de hidronefrosis. Se inició
hemodiálisis por insuficiencia renal aguda sobre crónica con hipervolemia e
hiperpotasemia.
PONENTE
Además de una herida progresiva en el pie, el paciente
ahora presenta disnea, hipotensión y lesión renal. La disnea puede ser
multifactorial y puede ser el resultado de anemia progresiva e hipervolemia;
Otras posibles causas, como embolia pulmonar o taponamiento cardíaco,
justifican una consideración urgente. La hipotensión surge por causas
cardiovasculares, distributivas o hipovolémicas. La lesión renal aguda puede
surgir por hipotensión de cualquier causa, incluida la sepsis (p. ej., una
infección que se origina en la úlcera del pie), o por lesión glomerular
progresiva, que se sugiere por la presencia de proteinuria. Los resultados de
la ecografía renal hacen que la lesión renal aguda posrenal sea poco probable.
La anemia inicial del paciente probablemente sea atribuible a la anemia de la
enfermedad renal crónica, y la progresión podría reflejar una hemorragia o hemólisis.
Aunque la hemólisis es poco probable sobre la base de los resultados de las
pruebas de laboratorio, un frotis de sangre periférica sería útil para
descartar microangiopatías trombóticas, como el síndrome urémico hemolítico o
la púrpura trombocitopénica trombótica, que pueden conducir a una lesión renal
aguda. Una úlcera cutánea, isquemia digital, lesión renal y (posible)
neuropatía son compatibles con una vasculopatía sistémica.
EVOLUCIÓN
La ecografía reveló una trombosis venosa profunda en
la vena femoral derecha. Se identificó una embolia pulmonar en una gammagrafía
de ventilación-perfusión. Se inició tratamiento con infusión de heparina y
warfarina. El empeoramiento de la insuficiencia renal del paciente se atribuyó
a su uso previo de fármacos antiinflamatorios no esteroideos. No se realizó
biopsia renal por uso de terapia anticoagulante. Al día siguiente, se
desarrollaron lesiones necróticas en los dedos de las manos, los pies, y el
pene. Se sospechó necrosis cutánea inducida por warfarina. Se suspendió el
tratamiento con warfarina y se pautó apixabán.
PONENTE
El desarrollo de trombosis venosa profunda y embolia
pulmonar en el contexto de una enfermedad subaguda puede indicar un cáncer
subyacente o vasculopatía autoinmune con hipercoagulabilidad asociada. Sin
embargo, la trombosis también podría ser el resultado de una enfermedad o
inflamación subaguda y una movilidad reducida por la ulceración del pie.
La necrosis cutánea inducida por warfarina es un
estado hipercoagulable transitorio inducido por el antagonismo de la vitamina K
y conduce a la oclusión vascular y la necrosis cutánea, pero este proceso suele
ocurrir después de unos días de tratamiento. Otras preocupaciones, dada la
aparición de lesiones necróticas a pesar del uso de anticoagulantes, incluyen
una forma grave subyacente de hipercoagulabilidad (p. ej., síndrome
antifosfolípido) o una vasculitis (p. ej., poliarteritis nodosa).
EVOLUCIÓN
Durante las siguientes 2 semanas, todas las heridas
necróticas permanecieron secas excepto la escara en el pie izquierdo, que se
volvió maloliente, con una mayor cantidad de drenaje y edema en el área
circundante. En un cultivo de heridas creció Pseudomonas aeruginosa . A pesar
del tratamiento con agentes antibióticos intravenosos, la cantidad de drenaje
de la herida y la profundidad de la herida aumentaron. El control urgente de la
fuente requirió una amputación debajo de la rodilla izquierda. No se realizaron
estudios angiográficos. Dos semanas después de la operación, fue dado de alta a
su casa con un plan para recibir cuidado de heridas y completar un ciclo de
apixabán de 3 meses. Se coordinó el seguimiento con su médico de atención
primaria y un especialista en medicina vascular.
Dos meses después del alta hospitalaria y 5 meses
después de la presentación inicial del paciente a su médico de atención
primaria, fue evaluado en una clínica de medicina vascular. Informó el
desarrollo gradual de múltiples heridas indoloras después del alta. Tenía una
gran escara que no cicatrizaba en la rodilla izquierda. Tres dedos del pie
derecho estaban necróticos y se había producido una autoamputación del segundo
dedo; también había una herida necrótica en el talón derecho. Tres dedos de la
mano derecha estaban necróticos, con preservación del pulgar y el quinto dígito,
y cuatro dedos de la mano izquierda estaban necróticos, con preservación del
pulgar ( Figura 1 ). Los pulsos radial y pedal no eran palpables.

Figura 1. Fotografías de heridas que no cicatrizan y
necróticas.
Se muestran fotografías clínicas de una herida que no
cicatriza cerca del área de la amputación por debajo de la rodilla izquierda
(Panel A), heridas necróticas en el talón y los dedos del pie derechos (Panel
B) y heridas necróticas en los dedos de ambas manos (Panel C).
PONENTE
Múltiples mecanismos pueden conducir a la isquemia de
órganos diana, que incluyen aterosclerosis, embolia, trombosis, disección,
vasoespasmo y vasculitis. Entre estas condiciones, la embolia, la trombosis y
la vasculitis son las más probables de ocurrir en esta distribución
generalizada durante un curso de tiempo subagudo.
No sería característico de una infección endovascular
afectar casi todos los dedos en ausencia de sepsis con coagulación
intravascular diseminada, y es poco probable que ocurra sepsis en el transcurso
de varios meses sin fiebre, leucocitosis o bacteriemia. Es concebible que un
proceso embólico no infeccioso, como la embolia de colesterol o el mixoma
auricular, haya lesionado tanto los riñones como los lechos vasculares
distales. Con la presencia de lesión renal grave y oclusión microvascular
difusa, se debe considerar la calcifilaxis (también conocida como
arteriolopatía urémica calcificante), aunque las lesiones cutáneas asociadas
suelen ser dolorosas, a diferencia de las que se desarrollaron en este paciente.
Junto con las evaluaciones de anemia, neuropatía, lesión renal e
hipercoagulabilidad, una ecocardiografía y un examen histopatológico de una
porción de tejido amputado de la pierna sería informativo.
EVOLUCIÓN
Después de un curso de 3 meses de apixabán, se realizó
una evaluación de trombofilia. No se detectó la mutación de protrombina 20210A;
la actividad de proteína C y proteína S fue 114% y 116%, respectivamente (rango
normal, 70 a 150); el nivel en sangre de homocisteína total fue de 10 μmol por litro
(rango normal, 5 a 15); no se detectó la mutación del factor V Leiden; el nivel
en sangre de lipoproteína (a) fue de 17 mg por decilitro (rango normal, 5 a
29); y la actividad de antitrombina III fue del 91% (rango normal, 80 a 130).
Prueba de anticoagulante lúpico, β 2-anticuerpos glicoproteína 1, anticuerpos
anticardiolipina, crioglobulinas, anticuerpos antinucleares y anticuerpos
anticitoplasma de neutrófilos perinucleares y citoplasmáticos fue negativo, al
igual que las pruebas de ADN de doble cadena, anti-Smith, anti-Scl-70, anti-ARN
polimerasa III, y anticuerpos anti-U1-ribonucleoproteína. Los niveles de
complemento eran normales. El tromboembolismo venoso se atribuyó al limitado
nivel de actividad del paciente en el contexto de lesiones gangrenosas.
Un ecocardiograma transtorácico no mostró masas ni
vegetaciones. El IDB y el ITB estaban
por encima del límite superior del rango normal, hallazgos que se atribuyeron a
arterias no comprimibles en las piernas y los pies. Las grabaciones de volumen
de pulso mostraron formas de onda monofásicas amortiguadas en todos los niveles
de ambas piernas debajo de los muslos. En la ecografía dúplex, se produjo una
interrupción difusa de las ondas sonoras como resultado de la sombra acústica
de la calcificación arterial. La angiografía por tomografía computarizada (TC)
del abdomen y las piernas reveló una extensa calcificación arterial en ambas
piernas sin evidencia de aterosclerosis ( Figura 2 ). Un mes después de la cita
de medicina vascular del paciente, hubo una expansión proximal significativa
del área necrótica de su pie derecho, y se sometió a una amputación debajo de
la rodilla derecha.

Figura 2. Angiografía por TC con Runoff.
El panel A muestra una imagen bidimensional de
proyección de máxima intensidad de ambas piernas (después de la amputación por
debajo de la rodilla izquierda). El panel B muestra una reconstrucción
tridimensional de ambas arterias femorales superficiales. El panel C muestra
una imagen bidimensional de las arterias poplítea derecha y debajo de la
rodilla. L denota izquierda y R derecha.
PONENTE
Las pruebas de ITB y IDB, la ecografía dúplex y la
angiografía por TC indican colectivamente una calcificación arterial grave
difusa. La enfermedad renal crónica y la diabetes lo ponen en riesgo de
enfermedad aterosclerótica, pero no se observó evidencia de aterosclerosis en
los estudios de imágenes.
Un trastorno que involucra calcificación arterial
generalizada y trombosis es la causa más probable de su isquemia y necrosis
generalizadas. Las pruebas serológicas autoinmunes negativas y los ensayos de
trombofilia hacen improbable una vasculitis autoinmune o una trombofilia
hereditaria.
EVOLUCIÓN
El examen histopatológico de una muestra quirúrgica de
la amputación debajo de la rodilla derecha reveló una calcificación arterial
medial densa sin depósito de calcio en las paredes capilares de la dermis o
subcutis (Figura 3 ) . No hubo evidencia de vasculitis, trombosis o
aterosclerosis. El nivel de calcio en sangre fue de 9,0 mg por decilitro (rango
normal, 8,5 a 10,2) y el nivel de fosfato de 4,3 mg por decilitro (rango
normal, 3,5 a 4,7). Los niveles de magnesio, 25-hidroxivitamina D y hormona
paratiroidea (PTH) eran normales.
Figura 3. Muestra de biopsia de la amputación por
debajo de la rodilla derecha.
La tinción con hematoxilina y eosina muestra
proliferación capilar reactiva (flecha) y calcificación medial en vasos de
paredes gruesas (punta de flecha). (Imagen cortesía de Ryanne A. Brown, MD).
PONENTE
La combinación de calcificación arterial medial
extensa, necrosis generalizada (posiblemente como resultado del uso de
warfarina) y enfermedad renal crónica avanzada es característica de la
calcifilaxis. Sin embargo, la presencia de heridas en tejido no adiposo, la
ausencia de dolor y los niveles normales de calcio, fósforo y PTH son atípicos.
Además, la pieza quirúrgica no muestra trombosis microvascular, una condición
que media parcialmente la necrosis asociada con la calcifilaxis, aunque la
trombosis puede ser dispersa y transitoria. Los pacientes con enfermedad renal
crónica avanzada y diabetes pueden tener calcificación de la arteria media que
es distinta de la calcificación de la íntima que ocurre con la aterosclerosis,
pero aún contribuye a la isquemia crítica de las extremidades.
EVOLUCIÓN
La necrosis progresiva de los dedos y la pérdida
adicional de extremidades continuaron, incluida la autoamputación de varios
dedos en cada mano. Dado que los hallazgos clínicos no eran consistentes con la
calcifilaxis, se buscó un diagnóstico alternativo. La secuenciación del genoma
completo del tejido arterial reveló una mutación de sentido erróneo en NT5E
(Fig. 4), que conduce a una deficiencia de CD73, una enzima que tiene un papel
en el metabolismo del fosfato y la calcificación tisular. El paciente recibió
un diagnóstico de calcificación arterial por deficiencia de CD73, también
conocido como ACDC.

Figura 4. La secuenciación del genoma completo
identifica una mutación sin sentido de adenina a guanina (c.1126AàG) en el gen
NT5E, que es coherente con las variaciones de nucleótido único identificadas
asociadas con enfermedades genéticas de calcificación arterial.
Fue tratado con cuidado de heridas e infusiones del
quelante de calcio tiosulfato de sodio durante la hemodiálisis sobre la base de
informes de beneficio de este agente en pacientes con calcificación vascular,
aunque no específicamente en aquellos con ACDC. Posteriormente, todas sus
heridas abiertas comenzaron a sanar y sus dedos no necróticos fueron salvados
(Fig. 5). Un año después, todas las heridas habían sanado y no habían aparecido
nuevas heridas. El paciente continuó recibiendo infusiones de tiosulfato de
sodio durante cada sesión de hemodiálisis.

Figura 5. Aspecto de las manos y piernas curadas.
Comentario
Este paciente presentó heridas periféricas e isquemia
de las extremidades que evolucionaron rápidamente hasta la pérdida de los dedos
y las extremidades. La identificación de calcificación vascular extensa sin
aterosclerosis replanteó el diagnóstico diferencial. Una vez descartada la calcifilaxis,
el estudio genómico condujo al diagnóstico de calcificación arterial por
deficiencia de CD73.
La arteriopatía periférica provoca un estrechamiento
luminal de las arterias, altera la perfusión de los brazos y las piernas y
puede conducir a una isquemia crítica de las extremidades. La arteriopatía
periférica es causada con mayor frecuencia por aterosclerosis y calcificación
de la íntima, que puede culminar en la ruptura de la placa y la trombosis. Los
pacientes con factores de riesgo de aterogénesis, como diabetes,
hiperlipidemia, hipertensión, tabaquismo y enfermedad renal crónica, son
especialmente susceptibles a la enfermedad arterial periférica y sus secuelas
clínicas. Sin embargo, cada vez se reconoce más que un mecanismo no
aterosclerótico, la calcificación de la arteria media, también puede causar la
oclusión de los vasos. 1
Los mecanismos moleculares que impulsan la
calcificación arterial medial son distintos de las vías que impulsan la
calcificación de la placa aterosclerótica, aunque ambos fenotipos están
asociados con eventos trombóticos. 1 Las consecuencias hemodinámicas de la
calcificación vascular derivan de una pérdida de elasticidad de los vasos
sanguíneos, 2 que puede conducir a isquemia, disfunción de órganos diana y
necrosis tisular. 3 En el paciente actual, los vasos sanguíneos no comprimibles
en las piernas, la sombra acústica en la ecografía dúplex y los hallazgos de la
angiografía por TC apuntaban a una calcificación vascular grave. El desarrollo
rápido y severo de las heridas sugirió que la enfermedad arterial periférica
del paciente difería de la calcificación arterial asociada a la aterosclerosis.
Una consideración temprana fue la calcifilaxis, un
trastorno raro que se caracteriza por calcificación vascular y se observa con
mayor frecuencia en pacientes con enfermedad renal terminal 4 y se asocia con
hiperparatiroidismo 5 , aunque se han informado casos en pacientes con función renal
normal y niveles normales de calcio, fosfato y PTH. Típicamente, las lesiones
cutáneas comienzan como un exantema violáceo y progresan a úlceras necróticas
dolorosas en áreas centrales de distribución alta de tejido adiposo. 5,6 Los
hallazgos histopatológicos incluyen calcificación de arterias de pequeño y
mediano calibre, depósito de calcio en las paredes capilares de la dermis y
subcutis y trombosis microvascular.
En este paciente, la ausencia de dolor; la
localización de heridas en manos, piernas y pies; y el nivel normal de PTH
llevó a los proveedores de salud a considerar causas alternativas. Además, la
evaluación histológica del espécimen quirúrgico de la amputación por debajo de
la rodilla derecha mostró una calcificación arterial medial densa de vasos de
paredes gruesas, pero no manifestaciones típicas de calcifilaxis, como depósito
de calcio o trombosis microvascular en la dermis o tejido adiposo subcutáneo.
La secuenciación del genoma completo identificó una
mutación sin sentido en NT5E (c.1126A→G, p.Thr376Ala), 7 una de varias
variantes de secuencia que se sabe que causan ACDC. NT5E codifica CD73, que
convierte el monofosfato de adenosina en adenosina y fosfato inorgánico. Las
mutaciones de pérdida de función que dan como resultado niveles bajos de CD73 conducen
a una disminución de la producción de adenosina y un aumento de la actividad de
la fosfatasa alcalina no específica de tejido, lo que resulta en una mayor
señalización de los mediadores aguas abajo que estimulan la calcificación
ectópica 8,9 ( Figura 6 ).
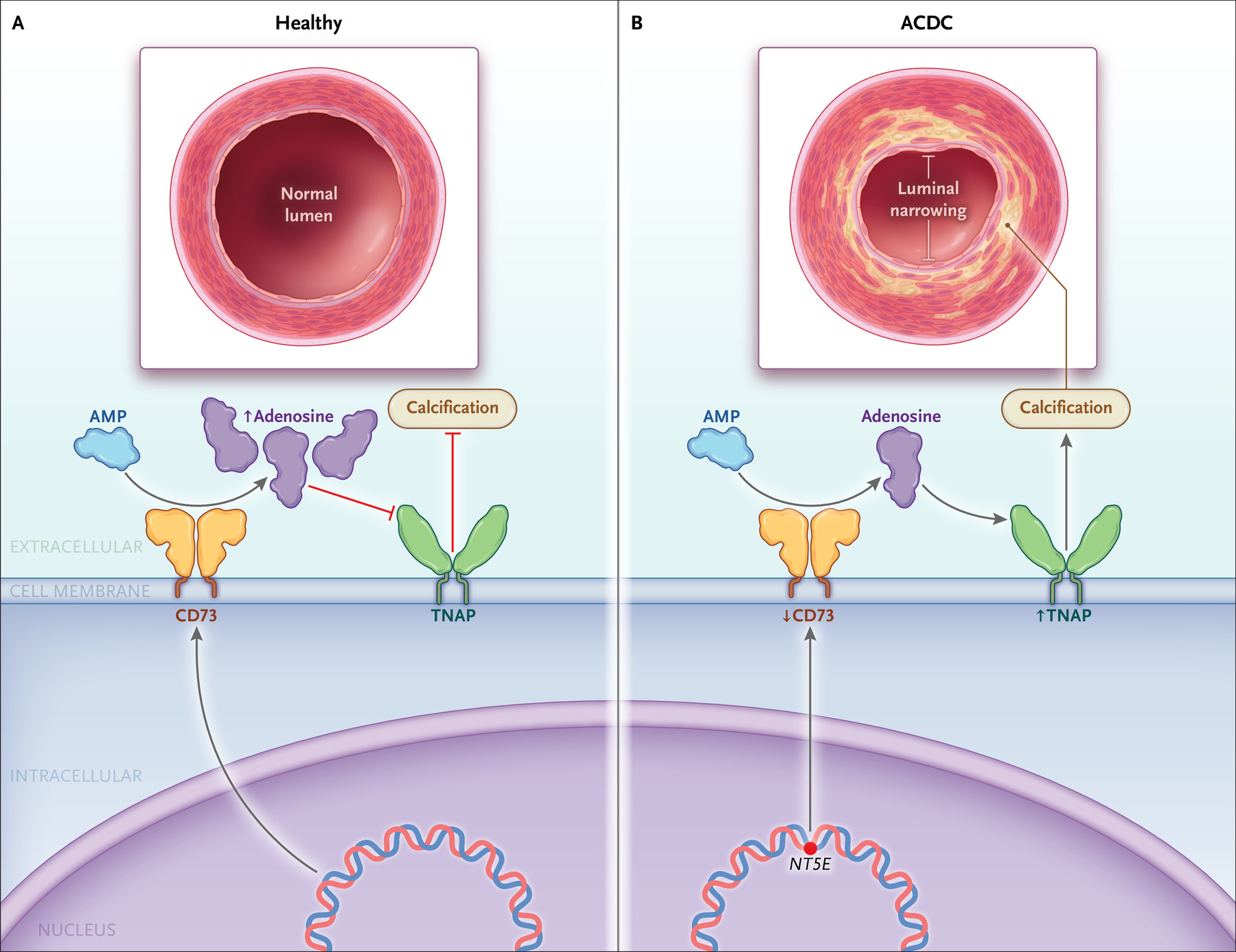
Figura 6. Calcificación ectópica en la calcificación
arterial por deficiencia de CD73 (ACDC).
Se muestra una representación de la cascada de
señalización que conduce a la calcificación ectópica en ACDC. Las flechas
muestran el cambio en los niveles de CD73 y de fosfatasa alcalina no específica
de tejido (TNAP) en ACDC. AMP denota monofosfato de adenosina.
El ACDC es un trastorno genético raro, autosómico
recesivo, de inicio en la edad adulta que causa calcificaciones periarticulares
y calcificación de las arterias periféricas (ilíaca, femoral y tibial) sin
afectar las arterias centrales más grandes, como la aorta y las arterias
carótida y coronaria. 10,11 El análisis histológico de tejido de pacientes con
ACDC revela calcificación de la capa arterial media que es distinta de la
calcificación de la íntima que acompaña a la aterosclerosis.
Las manifestaciones clínicas de la ACDC incluyen
artralgias en las manos y los pies, heridas, claudicación e isquemia crítica de
las extremidades. 10,12 En las primeras series de casos, se informó que la
mayoría de los pacientes tenían entre 20 y 50 años de edad y tenían función
renal normal, niveles normales de glucosa en sangre y niveles normales de
calcio, fosfato y PTH.
Este paciente tuvo una marcada mejoría en el proceso
isquémico después de que comenzó a recibir infusiones del tiosulfato de sodio
quelante de calcio. El subproducto de la reacción del tiosulfato de sodio con
el exceso de calcio se elimina fácilmente del cuerpo. Un estudio observacional
que involucró a pacientes con calcifilaxis mostró una menor incidencia de
muerte entre los que recibieron tiosulfato de sodio que la incidencia reportada
sobre la base de datos históricos entre pacientes que no habían recibido
tiosulfato de sodio, así como una reducción en el tamaño de la lesión asociada
con su uso . 13La eficacia del tiosulfato de sodio en este paciente sugiere que
los agentes que afectan el metabolismo del calcio pueden ser útiles en el
tratamiento de trastornos de calcificación rápidamente progresivos. Los
bisfosfonatos son inhibidores competitivos de la actividad de la fosfatasa
alcalina no específica de tejido y actualmente están bajo investigación como
tratamiento para ACDC (Número de ClinicalTrials.gov, NCT01585402. se abre en
una pestaña nueva).
El reconocimiento temprano de la enfermedad vascular
de este paciente que fue causada por un trastorno de calcificación vascular
podría haber llevado a la administración más temprana de tiosulfato de sodio y
puede haber resultado en la salvación de la extremidad. En el contexto de una
herida en una pierna o un pie, un resultado de IDB normal es insuficiente para
descartar enfermedad arterial periférica y debe combinarse con el uso de
registros de volumen de pulso y análisis de forma de onda, que pueden revelar
insuficiencia vascular. El desarrollo de lesiones gangrenosas durante la
hospitalización ameritó una evaluación angiográfica para investigar su causa y
asegurar una perfusión adecuada a las heridas recién desarrolladas.
Este caso destaca el hecho de que otras vías además de
la aterosclerosis pueden conducir a una isquemia crítica de las extremidades.
Para este paciente, la secuenciación de genes dirigió a los médicos a un
diagnóstico alternativo: ACDC.
Traducido de:
“An Alternate Explanation”
Tom Alsaigh, M.D., Gurpreet Dhaliwal, M.D., Eri
Fukaya, M.D., Ph.D., Nicholas J. Leeper, M.D., and Nazish Sayed, M.D., Ph.D.
The New England Journal of Medicine.
https://www.nejm.org/doi/full/10.1056/NEJMcps2210419?query=featured_home
References
1. St Hilaire C. Medial arterial calcification: a
significant and independent contributor of peripheral artery disease.
Arterioscler Thromb Vasc Biol 2022;42:253-60.
2. Demer LL, Tintut Y. Vascular calcification:
pathobiology of a multifaceted
disease. Circulation 2008;117:2938-48.
3. Giachelli CM. Vascular calcification
mechanisms. J Am Soc Nephrol 2004;15:
2959-64.
4. Yerram P, Chaudhary K. Calcific uremic
arteriolopathy in end stage renal disease: pathophysiology and management.
Ochsner J 2014;14:380-5.
5. Nigwekar SU, Thadhani R, Brandenburg VM.
Calciphylaxis. N Engl J Med
2018;378:1704-14.
6. Nigwekar SU, Zhao S, Wenger J, et al.
A nationally representative study of calcific uremic
arteriolopathy risk factors.
J Am Soc Nephrol 2016;27:3421-9.
7. National Center for Biotechnology Information.
ClinVar (VCV001175159.7), 2022
(https://www.ncbi.nlm.nih.gov/clinvar/
variation/1175159/?new_evidence=true).
8. Lanzer P, Boehm M, Sorribas V, et al.
Medial vascular calcification revisited: review and
perspectives. Eur Heart J 2014;
35:1515-25.
9. Jin H, St Hilaire C, Huang Y, et al. Increased
activity of TNAP compensates
for reduced adenosine production and
promotes ectopic calcification in the genetic disease
ACDC. Sci Signal 2016;9:
ra121.
10. St Hilaire C, Ziegler SG, Markello TC,
et al. NT5E mutations and arterial calcifications. N
Engl J Med 2011;364:432-
42.
11. Rutsch F, Buers I, Nitschke Y. Hereditary
disorders of cardiovascular calcification. Arterioscler Thromb Vasc Biol 2021;
41:35-47.
12. Cudrici CD, Newman KA, Ferrante EA,
et al. Multifocal calcific periarthritis with
distinctive clinical and radiological features in
patients with CD73 deficiency.
Rheumatology (Oxford) 2021;61:163-73.
13. Nigwekar SU, Brunelli SM, Meade D,
Wang W, Hymes J, Lacson E Jr. Sodium
thiosulfate therapy for calcific uremic arteriolopa