Presentación de caso
Un hombre de 60 años se presentó en este hospital
debido a fiebre, fatiga, artralgias, úlcera en la boca y rash cutáneo.
El paciente había estado en su estado de salud
habitual hasta 1 mes antes de la presentación actual, cuando desarrolló dolor
de garganta, fiebre y fatiga mientras visitaba a familiares en la región
suroeste de los Estados Unidos. Los síntomas persistieron a pesar del uso
regular de acetaminofén. Dos semanas antes de la presentación actual, el
paciente fue evaluado en el departamento de emergencias de un hospital local en
el suroeste, donde se informó que una prueba para el síndrome respiratorio
agudo severo coronavirus 2 (SARS-CoV-2) fue negativa. Se prescribió
amoxicilina-ácido clavulánico y doxiciclina por vía oral y se dio de alta a la
paciente.
Diez días antes de la presentación actual, se
desarrolló una úlcera en la lengua, junto con nódulos escamosos secos en la
cara y manos, y artralgias migratorias
en las muñecas, dedos, codos y hombros. Debido al empeoramiento de los
síntomas, el paciente regresó a su casa en Boston en avión. Cuando aterrizó en
Boston, el paciente se sentía demasiado débil para ponerse de pie y se llamó a
una ambulancia para transportarlo a este hospital.
En el servicio de urgencias, el paciente refirió
fiebres continuas, fatiga, artralgias y dolor de garganta. Había perdido 4,5 kg
durante un período de 1 mes. No tenía enrojecimiento ni dolor en los ojos,
úlceras genitales, dificultad para respirar, tos, náuseas, vómitos, diarrea o
dolor de cabeza. No había antecedentes médicos notables y el paciente no había
estado tomando ningún medicamento de forma regular antes de esta enfermedad.
Había completado un ciclo de amoxicilina-ácido clavulánico y doxiciclina por
vía oral el día antes de la presentación, y había estado tomando acetaminofén
durante el último mes. No se conocen alergias a medicamentos.
El paciente había nacido en China y había emigrado a
los Estados Unidos 20 años antes. Vivía con su esposa en Boston. No había
viajado internacionalmente durante más de 10 años. Había trabajado
anteriormente en mantenimiento ambiental pero estaba jubilado. No tenía
antecedentes de exposición a animales o agua dulce ni contactos conenfermos. No
había sido sexualmente activo durante varios años. Era un fumador con una
historia de 30 paquetes-año y bebía alcohol socialmente; no usaba drogas inyectables. Sus antecedentes familiares
incluían cáncer de colon en su padre, cáncer de pulmón en su madre, cáncer de
páncreas en su hermana y carcinoma hepatocelular en su hermano.
En el examen, la temperatura era de 37,8 ° C, la
presión arterial de 118/69 mm Hg, la frecuencia cardíaca de 78 latidos por
minuto, la frecuencia respiratoria de 20 respiraciones por minuto y la
saturación de oxígeno del 96% mientras el paciente respiraba aire ambiente. El
peso fue de 59 kg y el índice de masa corporal de 19,2. El paciente parecía indispuesto. No
había inyección conjuntival. Las membranas mucosas orales estaban secas. Había
una úlcera circular poco profunda en la cara lateral de la lengua en el lado
izquierdo, así como una placa blanca en la cara dorsal de la lengua. No había
adenopatías cervicales, axilares ni inguinales. El abdomen era blando e
indoloro, sin hepatoesplenomegalia. La fuerza fue de 4/5 en ambos hombros. El
resto del examen neurológico fue normal.Figura 1 ). No hubo lesiones genitales.



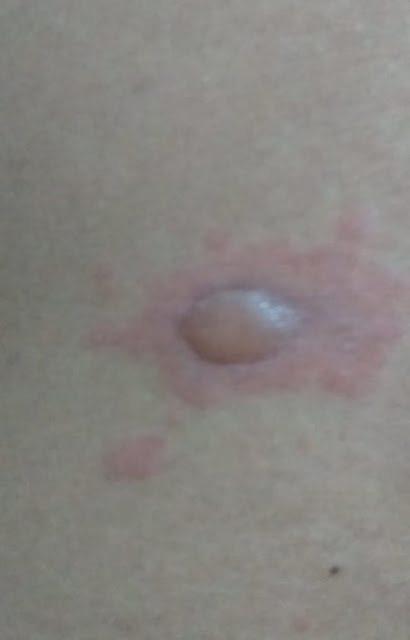
Figura 1. Fotografías clínicas.
Los paneles A y B muestran varios pequeños nódulos
escamosos con un borde de eritema parcheado en los dedos. El panel C muestra
áreas de hiperqueratosis en la cara lateral de los dedos. El panel D muestra
una erosión con costra serosanguínea en el codo izquierdo. El panel E muestra
dos nódulos eritematosos opacos y levemente violáceos en la frente. El panel F
muestra una úlcera circular poco profunda en la cara lateral de la lengua en el
lado izquierdo, así como placa blanca en la cara dorsal de la lengua.
La prueba de una muestra nasofaríngea para ARN del
SARS-CoV-2 fue negativa. El nivel de ferritina fue de 3089 μg por litro (rango de referencia,
20 a 300) y el nivel de creatina quinasa 505 U por litro (rango de referencia,
60 a 400). El análisis de orina mostró 2+ cetonas y 1+ proteína. Los resultados
de las pruebas de laboratorio adicionales se muestran en la Tabla 1 .




Tabla 1. Datos de laboratorio.
La radiografía de tórax realizada en el departamento
de emergencias reveló opacidades en parches bibasilares. La tomografía
computarizada (TC) de tórax, realizada tras la administración de contraste
intravenoso, reveló opacidades reticulares dependientes a lo largo de los
lóbulos inferiores de los pulmones ( fig. 2 ). La TC de abdomen y pelvis,
realizada tras la administración de contraste intravenoso, reveló un nódulo
suprarrenal izquierdo de 1,2 cm de diámetro.


Figura 2. Estudios de imagen.
Se realizó TC el día 1 de hospitalización. Imagen
axial a través de la parte inferior de los pulmones (Panel A) muestra opacidades
en vidrio esmerilado reticulares en
zonas declives, dependiente. Una imagen coronal a través de los campos pulmones
medios (Panel B) muestra engrosamiento perilobulillar y opacidades en vidrio
esmerilado y engrosamiento septal sin
panalización ni fibrosis. Una esonancia magnética se realizó el día 6 de
hospitalización. Una imagen oblicua del hombro izquierdo ponderado en T2 (Panel C) muestra parches leves de edema intramuscular que es
más pronunciado en el trapecio y deltoides. Se repitió la TC en el día 11 de hospital.
Una imagen axial a través de los campos pulmonares medios inferiores (Panel D) muestra opacidades en
vidrio esmerilado y consolidativas peribroncovasculares progresivas, un
hallazgo compatible con neumonía organizativa, así como pequeños derrames
pleurales izquierdos y trazas en el derecho. Imágenes coronales y sagitales
(paneles E y F, respectivamente) muestran engrosamiento perilobulillar y septal
y consolidación.
El paciente fue ingresado en el hospital. Se
administraron líquidos intravenosos y fluconazol oral. Se realizaron biopsias
de lesiones cutáneas en el codo izquierdo y lado izquierdo de la frente.
Durante los siguientes 5 días continuó la fatiga, artralgias, lesiones cutáneas
y dolor de boca, registrándose fiebres diarias con temperaturas de hasta 38,8 °
C. La lesión bucal no cedió con el uso de fluconazol, que se estaba
administrando para la candidiasis oral presunta, y se suspendió el fármaco.
El examen histopatológico de la muestra de biopsia
de la lesión de la frente reveló dermatitis de la interfase vacuolar con
hiperqueratosis, disqueratosis, incontinencia pigmentaria e inflamación crónica
perivascular escasa. Se observaron hallazgos similares en el examen
histopatológico de la muestra de biopsia de la lesión del codo.
El paciente desarrolló tos y aumentó el dolor en el hombro
izquierdo. En el quinto día de hospitalización, el nivel de creatina quinasa
fue de 906 U por litro. Otros resultados de pruebas de laboratorio se muestran
en la Tabla 1 . Al día siguiente, se obtuvieron estudios de imagen del hombro
izquierdo.
La resonancia magnética (RM) del hombro izquierdo,
realizada el día 6 de hospitalización, reveló un edema intramuscular leve en
parches que era más pronunciado en el trapecio y el deltoides, hallazgo
compatible con miositis ( fig. 2 ). No hubo colección de fluidos focalizada u
organizada.
El día 10 de hospitalización se realizó una biopsia
del deltoides izquierdo con el paciente bajo anestesia general. En las 24 horas
posteriores a la biopsia muscular, se desarrolló hipoxemia y el paciente
recibió 3 litros de oxígeno suplementario a través de una cánula nasal para
mantener una saturación de oxígeno superior al 93%. Al día siguiente, el
paciente recibió 8 litros de oxígeno suplementario a través de una cánula
nasal. Los resultados de las pruebas de laboratorio adicionales se muestran en
la Tabla 1 . Se obtuvieron estudios de imagen del tórax.
Una radiografía de tórax repetida reveló opacidades
en parches progresivas en las bases de los pulmones. La repetición de la TC de
tórax reveló opacidades en vidrio esmerilado peribroncovasculares y periféricas
progresivas y consolidativas que afectaban predominantemente a los lóbulos
inferiores, hallazgo compatible con neumonía organizativa ( fig. 2 ). Además,
estaban presentes nuevos derrames pleurales pequeños izquierdos y trazas en el lado
derecho.
Se realizó una prueba de diagnóstico.
DIAGNÓSTICO DIFERENCIAL
Este hombre de 60 años previamente sano presentó una
enfermedad multisistémica de rápida evolución. Varios aspectos de su
presentación, que incluyen fiebre, pérdida de peso, faringitis e inflamación
(con una velocidad de sedimentación globular elevada, nivel de proteína C
reactiva y nivel de ferritina), son hallazgos comunes de infección, cáncer,
enfermedad reumática y otras afecciones. Estos hallazgos son inespecíficos y no
ayudan a estrechar el diagnóstico diferencial. Las artralgias migratorias
también son inespecíficas, especialmente en ausencia de dolor a la palpación,
inflamación y calor de las articulaciones, características que podrían indicar
sinovitis. El título bajo de anticuerpos antinucleares (ANA) en un patrón
moteado es un resultado de prueba inespecífico que se puede observar en
personas sanas, así como en pacientes con enfermedad reumática, infección,
cáncer, así como en enfermedades autoinmunes órgano específicas como la
tiroiditis de Hashimoto.
Cuatro
hallazgos de la presentación de
este paciente tienen especificidad para ciertas enfermedades y, por lo tanto,
pueden ayudar a estrechar el diagnóstico diferencial. Primero, las pruebas de
laboratorio revelaron la presencia de anticuerpos anti-Ro. En segundo lugar, el
paciente tenía características de miositis, incluida la incapacidad para
ponerse de pie solo, debilidad en el hombro al examinarlo, evidencia de lesión
muscular en pruebas de laboratorio ( aspartato aminotransferasa y niveles de creatina quinasa elevados) y
edema muscular en la resonancia magnética. En tercer lugar, tenía varias manifestaciones
mucocutáneas de la enfermedad, incluida una úlcera en la lengua, así como
ulceraciones y nódulos en la cara y manos. La placa blanca de la lengua puede también
será una característica de su enfermedad subyacente. A pesar de que inicialmente
se consideró la candidiasis oral, la placa no cedió con la terapia con
fluconazol, y esta falta de respuesta sería atípica en un huésped presuntamente
inmunocompetente. En cuarto lugar, el paciente tenía una enfermedad pulmonar de
rápida evolución caracterizado por hipoxemia progresiva.
Dadas estas cuatro características en particular,
consideraría una enfermedad reumática en este paciente. Sin embargo, al evaluar
a un paciente por enfermedad reumática, también incluyo infecciones, cáncer y
reacciones a medicamentos en mi diagnóstico diferencial inicial. Estas
afecciones a menudo imitan una enfermedad reumática, pueden asociarse con el
desarrollo de enfermedad reumática (p. Ej., Poliarteritis nodosa asociada al
virus de la hepatitis B, dermatomiositis asociada al cáncer y lupus eritematoso
inducido por fármacos) y pueden exacerbarse o hacerse más difíciles de
diagnosticar después de la administración de glucocorticoides u otros
medicamentos inmunosupresores.
INFECCIÓN
En este paciente, el inicio rápido de la enfermedad,
los antecedentes de viaje al suroeste, la fiebre y los nódulos cutáneos podrían
sugerir una infección como la coccidioidomicosis. Sin embargo, una evaluación
exhaustiva de enfermedades infecciosas fue negativa.
CÁNCER
En este caso, se deben considerar el linfoma, la
leucemia, las discrasias de células plasmáticas y los cánceres de órganos
sólidos debido a la fiebre, la pérdida de peso, el nivel elevado de fosfatasa
alcalina, el nódulo suprarrenal, los antecedentes de tabaquismo y los
antecedentes familiares extensos de cáncer. Sin embargo, los recuentos
sanguíneos del paciente eran normales, no tenía linfadenopatía y las
características clave de su presentación, especialmente la miositis y el patrón
de enfermedad pulmonar, no son típicas del cáncer.
REACCIONES A MEDICAMENTOS
Aunque el paciente había estado expuesto a varios
agentes antimicrobianos, sus lesiones mucocutáneas no son típicas de una
reacción farmacológica. Su enfermedad comenzó antes de la exposición a
cualquier fármaco, y la enfermedad pulmonar y la miositis no serían
características esperadas de una reacción a un fármaco.
ENFERMEDADES REUMÁTICAS ASOCIADAS CON ANTICUERPOS
ANTI-RO
Debido a que la infección, el cáncer y las
reacciones a los medicamentos son diagnósticos poco probables en este caso, a
continuación consideraré cómo el desarrollo de anticuerpos anti-Ro, miositis,
lesiones mucocutáneas y enfermedad pulmonar rápidamente progresiva pueden
encajar mejor con un diagnóstico de enfermedad reumática. Las enfermedades
reumáticas más comunes asociadas con los anticuerpos anti-Ro son el síndrome de
Sjögren y el lupus eritematoso sistémico. Las enfermedades menos comunes
asociadas con los anticuerpos anti-Ro son la miositis inflamatoria, la
enfermedad mixta del tejido conectivo y la colangitis biliar primaria.
¿Podría el síndrome de Sjögren o el lupus explicar
la presentación de este paciente? El síndrome de Sjögren generalmente afecta a
mujeres en su sexta década y se caracteriza por un inicio insidioso de
síntomas, que incluyen ojos y boca secos. 1 Las características
extraglandulares pueden incluir síntomas constitucionales inespecíficos, dolor
y neuropatía. La miositis, la enfermedad pulmonar rápidamente progresiva y las
lesiones mucocutáneas observadas en este paciente serían atípicas del síndrome
de Sjögren, especialmente en ausencia de ojos y boca secos.
Al igual que el síndrome de Sjögren, el lupus a
menudo tiene una aparición insidiosa de síntomas y es más común en mujeres que
en hombres, aunque la enfermedad generalmente se desarrolla durante la edad
fértil. 2Las manifestaciones mucocutáneas son frecuentes, pero las lesiones
típicas observadas en pacientes con lupus (p. Ej., Lupus cutáneo agudo o
subagudo o lupus discoide) son distintas, en términos de distribución y
características, de los nódulos y ulceraciones descritos en este paciente;
tenía una úlcera en la lengua, a diferencia de las úlceras de la mucosa oral
que se ven a menudo en pacientes con lupus. Además, otras características
comunes del lupus, incluidas las citopenias, la presencia de autoanticuerpos
(p. Ej., Anticuerpos dirigidos contra el antígeno de Smith y el ADN
bicatenario) y la hipocomplementemia, estuvieron ausentes en este caso. Por lo
tanto, aunque el lupus y el síndrome de Sjögren se asocian comúnmente con anticuerpos
anti-Ro, ninguna de las dos enfermedades se adapta bien a la presentación de
este paciente.
ENFERMEDADES REUMÁTICAS ASOCIADAS CON MIOSITIS
En un enfoque para la evaluación de un paciente con
miositis, el primer paso es caracterizar la distribución de la afectación
muscular como simétrica o asimétrica. La piomiositis y el infarto muscular
diabético tienden a tener una distribución asimétrica, mientras que las
enfermedades reumáticas tienden a asociarse con miositis simétrica. La
excepción es la miositis por cuerpos de inclusión, que tiende a ser asimétrica.
En pacientes con miositis simétrica, como este
paciente, el siguiente paso en la evaluación es caracterizar la distribución
como principalmente proximal o distal. En las enfermedades reumáticas, a excepción
de la miositis por cuerpos de inclusión, tienden a estar afectados los músculos
proximales más que los distales. Las endocrinopatías, como el hipotiroidismo,
también pueden causar miopatía proximal simétrica, pero este paciente tenía un
nivel normal de tirotropina .
En general, la miositis proximal simétrica en este
paciente es compatible con miopatía inflamatoria idiopática, que se ha asociado
con anticuerpos anti-Ro. 3 Las categorías clásicas de miopatía inflamatoria
idiopática son miositis por cuerpos de inclusión, polimiositis, síndromes de
superposición (p. Ej., Síndrome de Sjögren y lupus), miositis necrotizante (p.
Ej., Miositis inducida por estatinas), síndrome antisintetasa y
dermatomiositis.
La miositis por cuerpos de inclusión es un diagnóstico
poco probable en este caso debido a la distribución de la afectación muscular.
La polimiositis es poco probable debido a las manifestaciones mucocutáneas. Los
síndromes de superposición son poco probables debido a la ausencia de rasgos
característicos de afecciones como el síndrome de Sjögren, el lupus, la
esclerosis sistémica y otras. El paciente no había sido tratado con estatinas y
no presentaba características de miositis necrotizante en la resonancia
magnética. Los pacientes con síndrome antisintetasa suelen tener miositis junto
con otras características clásicas, como el fenómeno de Raynaud, manos de
mecánico (hiperqueratosis en los lados de los dedos), 4 enfermedad pulmonar
intersticial, artritis y fiebre. 5Este paciente no tenía fenómeno de Raynaud ni
artritis inflamatoria, y sus lesiones cutáneas ulcerativas y nodulares no
serían características esperadas del síndrome antisintetasa; la rápida
progresión de su enfermedad pulmonar también sería atípica.
DERMATOMIOSITIS
¿Podría este paciente tener dermatomiositis? Las
manifestaciones cutáneas típicas de esta enfermedad incluyen pápulas de Gottron
(pápulas eritematosas en las articulaciones metacarpofalángicas e
interfalángicas proximales y otras superficies extensoras), erupción heliotropo
(una erupción eritematosa a violácea que a menudo involucra los párpados y el
área circundante), signo de la V (también conocido como signo del chal, una
erupción eritematosa en la parte anterior y posterior del torso) y el signo de
la cartuchera (una erupción eritematosa que afecta la zona lateral de la
cadera). Este paciente no tenía estas manifestaciones cutáneas típicas de la
dermatomiositis. Sin embargo, la dermatomiositis puede asociarse con enfermedad
pulmonar intersticial, que estaba presente en este paciente. Aunque la
dermatomiositis sigue siendo un posible diagnóstico en este caso, consideraré
otras enfermedades reumáticas que podrían explicar su presentación.
OTRAS ENFERMEDADES REUMÁTICAS ASOCIADAS CON MIOSITIS
La enfermedad de Still del adulto, la sarcoidosis y
la vasculitis pueden asociarse con miositis. Sin embargo, la miositis, la
enfermedad pulmonar y la presencia de anticuerpos anti-Ro no son
manifestaciones típicas de la enfermedad de Still del adulto, y en este
paciente no se presentó el exantema evanescente característico. 6 La
sarcoidosis afecta típicamente a pacientes más jóvenes y clásicamente causa
linfadenopatía hiliar, que estaba ausente en este paciente. 7 Además, lesiones
de la piel de este paciente y la presencia de anticuerpos anti-Ro no son
típicos de sarcoidosis.
La vasculitis es una categoría amplia de
enfermedades asociadas con la inflamación de las paredes de vasos pequeños,
medianos y grandes. 8 La poliarteritis nudosa es una consideración en este
paciente porque tenía antecedentes de hepatitis B, sobre la base de la prueba
positiva para anticuerpos del núcleo del virus de la hepatitis B. Los pacientes
con poliarteritis nudosa a menudo tienen mialgias, pero la miositis es menos
común y la enfermedad pulmonar observada en este paciente sería una
característica inusual de la poliarteritis nudosa.
La vasculitis por complejos inmunes, como IgA o
vasculitis crioglobulinémica, es un diagnóstico poco probable en este caso,
dados los niveles normales de C3 y C4. La vasculitis asociada a anticuerpos
anticitoplasma de neutrófilos (ANCA) también es poco probable, dado que una
prueba de ANCA fue negativa. Algunos pacientes con formas limitadas de
vasculitis asociada a ANCA (p. Ej., Enfermedad limitada a la cabeza y el
cuello) pueden tener una prueba de ANCA negativa, pero este paciente no tenía
otras características más típicas de la enfermedad. 9 Por último, la enfermedad
de Behçet puede asociarse a úlceras orales, pero la enfermedad pulmonar y la
presencia de anticuerpos anti-Ro no concuerdan con este diagnóstico.
¿Pueden los resultados de las biopsias de piel
ayudarnos a orientarnos hacia el diagnóstico más probable? Las muestras de
biopsia no mostraron características sugestivas de vasculitis de vasos pequeños
o medianos. No había granulomas, lo que sugeriría sarcoidosis. Había evidencia
de dermatitis de interfase, que típicamente se asocia con lupus o
dermatomiositis. Debido a que es poco probable que el lupus explique la
presentación de este paciente, revisaré la dermatomiositis como un posible diagnóstico.
RECONSIDERANDO LA DERMATOMIOSITIS
Otro enfoque para la evaluación de este paciente con
miopatía inflamatoria idiopática es considerar los fenotipos de miositis
asociados con varios anticuerpos específicos de miositis. 10Algunos anticuerpos
específicos de miositis (anticuerpos anti-factor intermediario transcripcional
1γ
[anti-TIF-1γ],
anticuerpos anti-Mi-2, anticuerpos anti-enzima activadora del modificador de
tipo ubiquitina pequeña [anti-SAE] y matriz antinuclear proteína 2 [anticuerpos
anti-NXP-2]) se asocian con dermatomiositis con manifestaciones cutáneas
típicas, pero estas manifestaciones no estaban presentes en este paciente. Otro
tipo (anticuerpos anti-histidil-transferencia de ARN sintetasa [anti-Jo-1]) se
asocia con el síndrome antisintetasa, pero el paciente no presentaba las
características típicas de esta afección, como artritis inflamatoria o fenómeno
de Raynaud.
Los anticuerpos anti-proteína 5 asociada a la
diferenciación del melanoma (anti-MDA-5) se asocian con dermatomiositis con
características cutáneas únicas, especialmente ulceraciones cutáneas y lesiones
nodulares, junto con enfermedad pulmonar intersticial rápidamente progresiva y
miopatía sutil; de hecho, muchos casos de dermatomiositis anti-MDA-5 son
amiopáticos. Las ulceraciones cutáneas que cubren las articulaciones
metacarpofalángicas e interfalángicas, los codos y los pliegues ungueales se
observan en hasta el 85% de los pacientes con dermatomiositis anti-MDA-5, a
menudo con hiperqueratosis digital lateral, que se observó en este paciente.
11,12Aproximadamente el 50% de los casos están asociados con úlceras orales, a
menudo en la lengua. Las artralgias, fiebre, presencia de anticuerpos anti-Ro,
resultados anormales en las pruebas de función hepática y niveles elevados de
ferritina observados en este paciente son comunes en pacientes con
dermatomiositis anti-MDA-5.
Sospecho que el diagnóstico más probable en este
paciente es dermatomiositis anti-MDA-5. Para establecer este diagnóstico,
revisaría los resultados de la biopsia muscular y la prueba de anticuerpos
dirigidos contra autoantígenos asociados a miositis, incluido MDA-5.
DIAGNÓSTICO PRESUNTIVO:
DERMATOMIOSITIS POR PROTEÍNA 5 ASOCIADA A
DIFERENCIACIÓN ANTI-MELANOMA (ANTI-MDA-5).
DISCUSIÓN PATOLÓGICA
La muestra de biopsia del deltoides izquierdo mostró
variación en el tamaño y la forma de las fibras musculares, con fibras
atróficas y anguladas dispersas. Las fibras atróficas tenían una distribución
preferencial en las zonas perifasciculares ( fig. 3 ). No había evidencia de
inflamación, vasculitis o fibras necróticas o en regeneración. La tinción
inmunohistoquímica de la muestra de biopsia en busca de marcadores
inflamatorios mostró solo células T positivas para CD3 escasas y macrófagos
perivasculares positivos para CD68 dispersos. La tinción inmunohistoquímica
para el complemento (C5b-9) fue negativa para los depósitos en las paredes
capilares. El examen microscópico electrónico reveló múltiples células
endoteliales que contenían inclusiones tubulorreticulares.

Figura 3. Muestra de biopsia del deltoides
izquierdo.
Las tinciones con hematoxilina y eosina de cortes de
tejido congelado muestran músculo esquelético con atrofia perifascicular (Panel
A, flechas) y variación en el tamaño y la forma de las fibras musculares, con
fibras anguladas y atróficas dispersas (Panel B, asteriscos). Tinciones inmunohistoquímicas
de los cortes de tejido fijado con formalina muestran muy pocos signos de
inflamación crónica, incluidas escasas células T CD3 positivas (Panel C) y macrófagos
perivasculares positivos para CD68 dispersos (Panel D). Las imágenes
microscópicas electrónicas muestran células endoteliales con inclusiones tubulorreticulares
(Paneles E y F, asteriscos).
A pesar de la ausencia de inflamación y lesión
activa de los miocitos, la presencia de atrofia perifascicular e inclusiones
endoteliales en el músculo, en combinación con los hallazgos
dermatopatológicos, es consistente con el diagnóstico de dermatomiositis. 13 La
ausencia de enfermedad activa podría deberse al muestreo porque la inflamación
puede ser irregular o la enfermedad puede estar en una fase inactiva.
Se utilizó un ensayo inmunoabsorbente ligado a
enzimas (ELISA) para detectar autoanticuerpos asociados a miositis. El suero
del paciente contenía anticuerpos dirigidos contra MDA-5 y Ro-52 ( Fig. 4 ).
Ambas proteínas tienen funciones importantes en la inmunidad celular innata.
MDA-5 es una proteína citoplasmática que es capaz de detectar el ARN viral de
doble hebra y posteriormente contribuye a la inmunidad antiviral regulando positivamente
los genes estimulados por interferón. 14 Ro-52 es una ubiquitina ligasa E3 que
se une a virus que han sido recubiertos con anticuerpos y causa la degradación
de las proteínas de la cápside. La pérdida de las proteínas de la cápside viral
conduce a la liberación prematura de ácidos nucleicos virales, que luego pueden
ser detectados por proteínas como MDA-5. 15 Se desconocen las funciones
potenciales de MDA-5 y Ro-52 en la patogenia de la dermatomiositis.

Figura 4. Resultados de la prueba de autoanticuerpos
asociados a miositis.
El panel A muestra los resultados de un ensayo
inmunoabsorbente ligado a enzimas utilizado para detectar autoanticuerpos
asociados a miositis. El ensayo consiste de una tira de prueba con 16
autoantígenos purificados dispuestos en bandas paralelas. La banda 17 es un
control positivo (inmunoglobulina humana) para la reacción colorimétrica.
Después de la incubación con suero de paciente diluido, se detectaron
anticuerpos que se adhirieron a la tira reactiva con el uso de anticuerpos IgG
antihumanos marcados con enzimas. El suero de control positivo contenía anticuerpos
dirigidos contra NXP-2. El suero del paciente contenía anticuerpos que
reaccionaban con una banda ligeramente más alta que la banda NXP-2, que correspondía
a la ubicación de MDA-5. El suero del paciente también contenía anticuerpos que
reaccionaban con Ro-52. Los paneles B a G muestran los resultados de un ensayo
de inmunofluorescencia indirecta utilizado para confirmar la presencia de anticuerpos
anti – MDA-5 y anti– Ro-52 en el suero del paciente. Para aumentar los niveles
de estos autoantígenos en el sustrato de células HEp-2, las células HEp-2
fueron transitoriamente transfectados por plásmidos que codifican la proteína
verde fluorescente (GFP) fusionados con MDA-5 (Paneles B, C y D) o Ro-52
(Paneles E, F y G). Se utilizó anticuerpo monoclonal anti-GFP para identificar
las células transfectadas con éxito (paneles B y E, en verde). Los anticuerpos
en el suero del paciente tiñeron solo el citoplasma de las células que
expresaban las proteínas GFP-MDA-5 o GFP-Ro-52 (paneles C y F, en rojo), un
hallazgo que confirmó la presencia de autoanticuerpos. El suero del paciente no
contenía anticuerpos dirigidos contra GFP solo (no se muestra). Tinción con 4
′, se utilizó diclorhidrato de 6-diamidina-2-fenilindol (DAPI) para mostrar la
ubicación de los núcleos (paneles D y G, en azul) en todas las células, incluidos
los que no expresaban las proteínas GFP-MDA-5 o GFP-Ro-52. Las flechas indican
células que expresaron proteínas de fusión GFP.
Un estudio reciente mostró la prevalencia de
resultados falsos positivos asociados con algunos ensayos disponibles
comercialmente que se utilizan para detectar autoanticuerpos asociados a
miositis. Por tanto, se utilizaron inmunofluorescencia indirecta y sustrato de
células HEp-2 para confirmar la presencia de autoanticuerpos contra MDA-5 y
Ro-52 en el suero del paciente. Las células HEp-2 no expresan MDA-5 y expresan
niveles muy bajos de Ro-52, lo que explica el bajo título de ANA en este paciente.
Para aumentar los niveles de estos autoantígenos en el sustrato de la célula
HEp-2, las células HEp-2 fueron transfectadas transitoriamente por plásmidos
que codifican la proteína verde fluorescente (GFP) fusionada con MDA-5 o Ro-52.
Posteriormente, las células se fijaron, se permeabilizaron y se tiñeron con el
suero del paciente. Los anticuerpos en el suero del paciente tiñeron solo las
células que expresaban GFP-MDA-5 o GFP-Ro-52 ( Fig. 4). En conjunto, los
resultados del ELISA y el ensayo de inmunofluorescencia indirecta confirmaron
la presencia de anticuerpos anti-MDA-5 y anti-Ro-52 en este paciente.
DIAGNÓSTICO PATOLÓGICO
DERMATOMIOSITIS POR PROTEÍNA 5 ASOCIADA A
DIFERENCIACIÓN ANTI-MELANOMA (ANTI-MDA-5).
DISCUSIÓN DEL MANEJO
Consideramos muchas opciones de tratamiento para
este paciente, incluida la terapia con glucocorticoides en dosis altas,
ciclofosfamida, rituximab, inhibidores de calcineurina, inmunoglobulina
intravenosa, plasmaféresis y tofacitinib. Algunos pacientes con dermatomiositis
anti-MDA-5 tienen enfermedad pulmonar intersticial rápidamente progresiva y
este paciente tenía varios factores de riesgo de progresión rápida, incluido el
nivel alto de ferritina, la presencia de anticuerpos anti-Ro-52 y el origen
étnico asiático. 17,18 Hemos tenido en cuenta estos factores al sopesar el
riesgo de muerte por enfermedad pulmonar intersticial rápidamente progresiva
frente al riesgo potencial de infecciones oportunistas asociadas con la
inmunosupresión.
Los datos limitados de los análisis de casos y
controles históricos mostraron que la terapia de combinación con dosis altas de
glucocorticoides, tacrolimus y ciclofosfamida intravenosa se asoció con una
tasa de supervivencia a los 6 meses del 89% en el grupo de tratamiento, en comparación
con una tasa del 33% en el historial. controles que recibieron estas terapias
en una adición secuencial. 19 Sobre la base de estos datos, iniciamos estos
tres fármacos inmunosupresores, además de la inmunoglobulina intravenosa, el
día 14 de hospitalización.
A pesar de este tratamiento, el estado del paciente
continuó empeorando y requirió oxigenoterapia a través de una cánula nasal de
alto flujo. El día 28 de hospitalización, el paciente comenzó a recibir
ventilación mecánica. El tratamiento con tofacitinib, un inhibidor oral de las
cinasas Janus 1 y 3, se inició como terapia de rescate el día 34 de
hospitalización. 20 Sin embargo, al día siguiente se produjo una insuficiencia
renal. Se consideró la hemodiálisis con oxigenación por membrana extracorpórea
venovenosa como puente al trasplante de pulmón. Sin embargo, debido a su
historial de tabaquismo, el paciente no fue considerado candidato a trasplante
de pulmón. El día 37 de hospitalización, después de discusiones adicionales con
la familia del paciente sobre su mal pronóstico, los objetivos de la atención
se trasladaron a medidas de comodidad únicamente, y el paciente falleció el
mismo día.
DIAGNOSTICO FINAL
DERMATOMIOSITIS POR PROTEÍNA 5 ASOCIADA A
DIFERENCIACIÓN ANTI-MELANOMA (ANTI-MDA-5).
Traducción de:
Zachary S.
Wallace, M.D., Karen Rodriguez, M.D., Jonathan Dau, M.D., Donald B. Bloch,
M.D., and Samantha N. Champion, M.D.
https://www.nejm.org/doi/full/10.1056/NEJMcpc2107353?query=featured_home
REFERENCIAS
1. Mariette X,
Criswell LA. Primary
Sjögren’s
syndrome. N Engl J Med 2018;
378: 931-9.
2. Lisnevskaia
L, Murphy G, Isenberg D.
Systemic lupus
erythematosus. Lancet
2014; 384:
1878-88.
3. Dimachkie MM,
Barohn RJ. Inclusion
body myositis.
Neurol Clin 2014; 32: 629-
46.
4. Concha JSS, Merola JF, Fiorentino D,
Werth VP.
Re-examining mechanic’s
hands as a
characteristic skin finding in
dermatomyositis.
J Am Acad Dermatol
2018; 78(4):
769-775.e2.
5. Marco JL,
Collins BF. Clinical manifestations
and treatment of
antisynthetase
syndrome. Best
Pract Res Clin Rheumatol
2020; 34:
101503.
6. Feist E,
Mitrovic S, Fautrel B. Mechanisms,
biomarkers and
targets for adultonset
Still’s disease.
Nat Rev Rheumatol
2018; 14:
603-18.
7. Valeyre D,
Prasse A, Nunes H, Uzunhan
Y, Brillet P-Y, Müller-Quernheim J.
Sarcoidosis.
Lancet 2014; 383: 1155-67.
8. Jennette JC,
Falk RJ, Bacon PA, et al.
2012 revised
International Chapel Hill
Consensus
Conference Nomenclature of
Vasculitides.
Arthritis Rheum 2013; 65:
1-11.
9. Finkielman
JD, Lee AS, Hummel AM,
et al. ANCA are
detectable in nearly all
patients with
active severe Wegener’s
granulomatosis.
Am J Med 2007; 120(7):
643.e9-643.e14.
10. Riddell V,
Bagby S, McHugh N. Myositis
autoantibodies:
recent perspectives.
Curr Opin
Rheumatol 2020; 32: 548-52.
11. Kurtzman
DJB, Vleugels RA. Antimelanoma
differentiation-associated
gene
5 (MDA5)
dermatomyositis: a concise review
with an emphasis
on distinctive
clinical
features. J Am Acad Dermatol
2018; 78:
776-85.
12. Narang NS,
Casciola-Rosen L, Li S,
Chung L,
Fiorentino DF. Cutaneous ulceration
in
dermatomyositis: association with
anti-melanoma
differentiation-associated
gene 5
antibodies and interstitial lung
disease.
Arthritis Care Res (Hoboken)
2015; 67:
667-72.
13. Vattemi G,
Mirabella M, Guglielmi V,
et al. Muscle
biopsy features of idiopathic
inflammatory
myopathies and differential
diagnosis. Auto
Immun Highlights
2014; 5: 77-85.
14. Brisse M, Ly
H. Comparative structure
and function
analysis of the RIG-I-like receptors:
RIG-I and MDA5.
Front Immunol
2019; 10: 1586.
15. Watkinson
RE, McEwan WA, Tam
JCH, Vaysburd M,
James LC. TRIM21 promotes
cGAS and RIG-I
sensing of viral
genomes during
infection by antibodyopsonized
Virus. PLoS
Pathog 2015; 11(10):
e1005253.
16. Tansley SL,
Li D, Betteridge ZE,
McHugh NJ. The
reliability of immunoassays
to detect
autoantibodies in patients
with myositis is
dependent on autoantibody
specificity.
Rheumatology (Oxford)
2020; 59:
2109-14.
17. Huang K,
Vinik O, Shojania K, et al.
Clinical
spectrum and therapeutics in
Canadian
patients with anti-melanoma
differentiation-associated
gene 5 (MDA5)-
positive
dermatomyositis: a case-based
review.
Rheumatol Int 2019; 39: 1971-81.
18. Moghadam-Kia
S, Oddis CV, Aggarwal
R. Anti-MDA5
antibody spectrum in
western world.
Curr Rheumatol Rep 2018;
20: 78.
19. Tsuji H, Nakashima R, Hosono Y, et al.
Multicenter
prospective study of the efficacy
and safety of
combined immunosuppressive
therapy with
high-dose glucocorticoid,
tacrolimus, and
cyclophosphamide
in interstitial
lung diseases accompanied
by anti-melanoma
differentiation-associated
gene 5-positive
dermatomyositis.
Arthritis
Rheumatol 2020; 72: 488-98.
20. Kurasawa K,
Arai S, Namiki Y, et al.
Tofacitinib for
refractory interstitial lung
diseases in
anti-melanoma differentiation-
associated 5
gene antibody-positive
dermatomyositis.
Rheumatology (Oxford)
2018; 57:
2114-9.