Las enfermedades renales parenquimatosas se pueden considerar anatómicamente bajo los títulos de enfermedad glomerular, tubular, tubulointersticial y vascular. La mayoría de los pacientes presentan un síndrome clínico de lesión de la nefrona. Otras manifestaciones del síndrome nefrótico dependen de cómo esté afectado el riñón; por ejemplo, la glomerulonefritis (GN) se presenta con empeoramiento de la función renal, hipertensión, hematuria, proteinuria y cilindros de glóbulos rojos. La nefritis tubulointersticial se presenta con azotemia, piuria y/o cilindros de glóbulos blancos.
SÍNDROMES DE LESIÓN NEFRONAL
HEMATURIA GLOMERULAR AISLADA
La hematuria glomerular aislada se define como
hematuria microscópica persistente con glóbulos rojos dismórficos (GR), “tira
reactiva” negativa para proteinuria, concentración sérica de creatinina normal
y presión arterial normal. Las causas comunes incluyen nefropatía por IgA, nefritis
hereditaria y enfermedad de la membrana basal delgada (fig. 63.1).

Figura. 63. 1. Estudio de hematuria. ANA, Anticuerpo
antinuclear; ANCA, anticuerpo anticitoplasma de neutrófilo; Anti-GBM, membrana
basal antiglomerular; CBC, hemograma completo; Dx, diagnóstico; IgA,
inmunoglobulina A; PCR: relación proteína creatinina; PT, tiempo de
protrombina; PTT, tiempo parcial de tromboplastina; RBC/hpf, glóbulos
rojos/campo de alta potencia; Rx, reacción; ITU, infección del tracto urinario.
PROTEINURIA NO NEFRÓTICA AISLADA
La proteinuria no nefrótica aislada se define como
proteinuria >150 mg/dl (60% de la cual suele ser albuminuria). La figura
63.2 muestra las causas de la proteinuria persistente y funcional.

Fig. 63. 2. Estudio de proteinuria.
Una biopsia renal rara vez está indicada en pacientes
con proteinuria de bajo grado (<500-1000 mg/d) si no hay hematuria, ausencia
de evidencia clínica o serológica de enfermedad sistémica que pueda causar GN y
función renal normal (ver Figura 63.2).
SÍNDROME NEFRÓTICO
El síndrome nefrótico se define como proteinuria
intensa (≥3,5 g/d/1,73 m2 de superficie), edema, hipoalbuminemia e
hiperlipidemia.
Las causas del síndrome nefrótico se muestran en el
cuadro 63.1.

SÍNDROME NEFRÍTICO AGUDO
Se caracteriza por hematuria, cilindros de glóbulos
rojos, azotemia, proteinuria variable, oliguria, edema e hipertensión. A menudo
es causada por una enfermedad sistémica que requiere una biopsia renal para
establecer su diagnóstico y orientar el tratamiento. El ejemplo clásico de esto
es la GN posinfecciosa aguda (PIGN). Los ejemplos incluyen lupus eritematoso
sistémico (SLE), poliangeítis microscópica, granulomatosis de Wegener (granulomatosis
con poliangeítis) y enfermedad de la membrana basal antiglomerular (anti-GBM).
LESIÓN RENAL AGUDA SIN EXPLICACIÓN
La mayoría de las veces, el diagnóstico no se basa en
una biopsia renal. La biopsia está indicada en aquellas situaciones en las que el
diagnóstico es incierto, como puede ser en ocasiones el caso de la nefritis
intersticial aguda secundaria a fármacos.
CAUSAS IMPORTANTES DEL SÍNDROME NEFRÓTICO
ENFERMEDAD DE CAMBIOS MÍNIMOS
La enfermedad de cambios mínimos (MCD, por sus siglas
en inglés) es una de las principales causas del síndrome nefrótico en niños y
representa del 15 al 20 % de los casos en adultos. La causa subyacente exacta
de la MCD no está clara. La evidencia acumulada sugiere que la disfunción
sistémica de las células T da como resultado la producción de un factor de
permeabilidad circulante o una anomalía en las citocinas. La interleucina
(IL)-13, una citocina h2 antiinflamatoria, está implicada. Estos factores circulantes
afectan directamente a los podocitos, lo que da como resultado un borramiento
del proceso del pie y una marcada proteinuria. Aunque la MCD idiopática
(primaria) es la forma más común, las formas secundarias de MCD ocurren en
varios entornos que incluyen la enfermedad de Hodgkin y otros trastornos
linfoproliferativos, respuesta alérgica (picadura de abeja, inmunización,
medicamentos como los antiinflamatorios no esteroideos) e infecciones (VIH) .
El inicio repentino de un síndrome nefrótico
(proteinuria glomerular >3.5 g/día en un adulto o >40 mg/h/m2 en un niño,
hipoalbuminemia y edema) es la presentación típica de la MCD. La hematuria y/o
la hipertensión pueden estar presentes en alrededor del 20% de los casos. La
función renal (evaluada por la creatinina sérica o la tasa de filtración
glomerular estimada [eGFR]) suele ser normal, pero entre el 15 % y el 30 % de
los adultos (generalmente mayores de 40 años) pueden presentar o desarrollar
lesión renal aguda (AKI). La patología renal se caracteriza por anomalías
glomerulares mínimas o ausentes en el microscopio óptico o inmunofluorescente
(fig. 63.3).
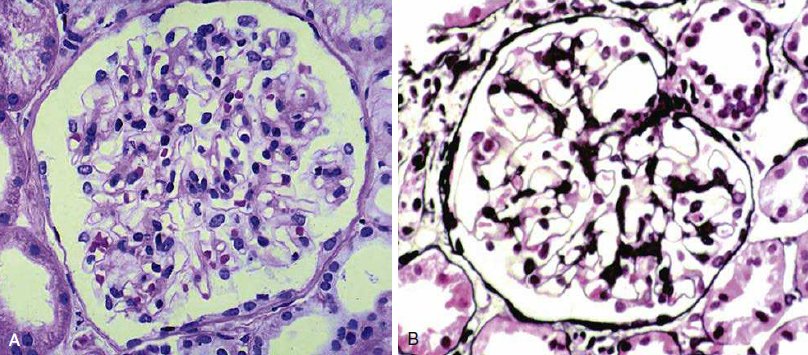
Figura 63. 3. Comparación normal (A) versus
enfermedad de cambios mínimos (B).
La observación más consistente se ve en la microscopía
electrónica: simplificación de las células epiteliales viscerales con
borramiento generalizado y difuso de los procesos podocitarios. El tratamiento
consiste en una prueba de prednisona a 1 mg/kg/día. La respuesta a los
esteroides suele ser espectacular.
El tratamiento en niños consiste en prednisona 60
mg/m2/d (dosis máxima de 80 mg/m2/d) hasta que se haya inducido una remisión (o
durante 4 semanas, lo que sea más corto) y luego 35 a 40 mg/m2 en días alternos
durante unas 12 semanas seguidas de una disminución gradual. Los adultos con
MCD establecida se tratan con 1 mg/kg/día de prednisona hasta la remisión (o
durante 6 semanas, lo que sea más corto), seguido de una disminución gradual.
Por lo general, el tratamiento se continúa durante aproximadamente 8 semanas en
niños y 16 semanas en adultos, con una disminución gradual a partir de
entonces. Cerca del 95 % de los niños y alrededor del 90 % de los adultos
responderán con una remisión completa de la proteinuria con este régimen
inicial, más temprano en los niños y más tarde en los adultos. Sin embargo,
muchos pacientes (40%–60%) recaerán durante la fase de disminución del
tratamiento (recaídas dependientes de esteroides) o semanas, meses o incluso
años después. Las infecciones o alergias intercurrentes pueden desencadenar una
recaída. Algunos pacientes tienen recaídas frecuentes (>2 años) y requieren
tratamientos repetidos.
GLOMERULOESCLEROSIS FOCAL Y SEGMENTARIA
La glomeruloesclerosis focal y segmentaria (GEFS) es
un patrón de lesión que resulta de la lesión de los podocitos glomerulares. La
categoría amplia incluye formas primarias y secundarias. La GEFS primaria es un
diagnóstico clinicopatológico caracterizado por la ausencia de evidencia
clínica o histológica de un antecedente de GN, depósito de inmunocomplejos o
enfermedad sistémica con afectación glomerular. La forma primaria de GEFS es
causada por una lesión podocitaria de origen desconocido. Las formas
secundarias de GEFS pueden ser causadas por anomalías familiares/genéticas
(mutaciones de α-actinina-4), inducidas por fármacos (heroína, pamidronato),
respuestas estructurales y funcionales adaptativas del glomérulo (en el
contexto de una masa reducida de nefronas, como la agenesia renal). ,
displasia, necrosis cortical, oligomeganefronia) o masa normal de nefronas (p.
ej., obesidad, hipertensión), o virus (p. ej., VIH).
La presentación clínica típica es con el inicio
insidioso de proteinuria no nefrótica o síndrome nefrótico. La hipertensión y
la insuficiencia renal son comunes en el momento de la presentación. La
excreción urinaria de proteínas puede ser muy alta, a veces ≥20 g/día, y la
proteinuria no es selectiva con una excreción fraccional alta de IgG (a menudo
>0.2). Los componentes del complemento sérico son normales.
Es importante diferenciar las formas primarias de las
secundarias de GEFS. Una historia de proteinuria en rango subnefrótico, un
inicio más lento de los síntomas y factores predisponentes acompañados de
hallazgos típicos de biopsia son útiles para hacer el diagnóstico de GEFS
secundaria.
Los hallazgos de la biopsia renal de la GEFS son
variables. Hay varias variantes histológicas: colapsante, tip, celular,
perihiliar y GEFS (NOS). En la FSGS (no especificada [NOS]) hay al menos un
glomérulo con expansión segmentaria de la matriz con obliteración de la luz capilar
(fig. 63.4).

Figura 63. 4. Glomeruloesclerosis segmentaria focal.
La variante colapsante, que se observa en la
nefropatía relacionada con el VIH, es menos común y muestra el colapso del
segmento o de todo el penacho glomerular con hiperplasia e hipertrofia de los
podocitos suprayacentes. En la variante de punta, la microscopía electrónica es
útil para diferenciar formas primarias y secundarias de GEFS. En las formas
primarias de FSGS, además de las anomalías en el microscopio óptico, existen
anomalías distintas en el microscopio electrónico, borramiento difuso de los
pies de las células epiteliales incluso en áreas donde no hay anomalías en el
microscopio óptico y microscopía electrónica en ausencia de degeneración de
microvellosidades. , y desprendimiento focal de los pies de los GBM. La GEFS secundaria se caracteriza por la preservación relativa de los procesos
podocitarios en los glomérulos no esclerosados.
A diferencia de MCD, GEFS generalmente es resistente a
la terapia con esteroides. Si los esteroides se usan solos, se administra
prednisona oral en una dosis de 1 mg/kg/d o 2 mg/kg en días alternos durante 2
a 3 meses con disminución gradual durante otros 2 a 3 meses. Sin embargo, sólo
alrededor de la mitad de los pacientes responden al tratamiento con esteroides
y con frecuencia los pacientes necesitan un tratamiento complementario, como
ciclosporina (4 a 5 mg/kg/día durante tres a seis meses).
Acthar gel (hormona adrenocorticotrópica, inyección de
corticotropina de depósito) ha demostrado ser eficaz en una variedad de
situaciones de síndrome nefrótico refractario, incluso en pacientes con GEFS
resistente. Desafortunadamente, la evidencia se basa en gran medida en datos de
observación y el tratamiento es costoso (Madan et al., 2016; Tune et al., 1997).
Entre los niños, ofatumumab, un anticuerpo monoclonal anti-CD20 humanizado, se
ha mostrado prometedor, aunque se necesitarán estudios más amplios para sacar
conclusiones definitivas. La resistencia al tratamiento (ausencia de una
remisión completa o parcial) está fuertemente asociada con un alto riesgo de
progresión a enfermedad renal en etapa terminal (ESRD), que puede ser muy
rápida si la proteinuria es >15 a 20 g/d. Los pacientes con FSGS pueden
recaer después de una remisión completa o parcial. También se presenta una
mayor incidencia de recurrencia después del trasplante.
GLOMERULOPATÍA MEMBRANOSA
La nefropatía membranosa (MN) es una de las causas más
comunes del síndrome nefrótico en adultos no diabéticos mayores de 40 años. La
MN puede ser primaria/idiopática (75%–85% de los adultos). Solo alrededor del
25% de los niños con MN tienen la forma idiopática.
La naturaleza autoinmune de la MN fue demostrada por
Beck et al. en 2009 con la identificación de la IgG depositada en el glomerular
(un autoanticuerpo predominantemente IgG4, anti-PLA2R) contra el receptor de la
fosfolipasa A2 (PLA2R) en el podocito glomerular en ≈70% de los pacientes con
MN primaria. El anticuerpo anti-PLA2R se puede ensayar y utilizar en el
diagnóstico y seguimiento del tratamiento de pacientes con MN. En 2014, se
identificó un segundo autoantígeno, el dominio 7A que contiene la trombospondina
tipo 1.
La MN puede ser secundaria a otras enfermedades como
la antigenemia de la hepatitis B, las enfermedades autoinmunes, la tiroiditis,
las neoplasias malignas, el uso de ciertos fármacos como el oro, la
penicilamina, el captopril y los antiinflamatorios no esteroideos, y más
recientemente se ha asociado a enfermedades relacionadas con la IgG4.
MN puede afectar a todos los grupos de edad, pero
tiene una incidencia máxima en los años 40 y 50. No tiene predilección racial.
En la presentación, 60% a 70% de los pacientes tienen síndrome nefrótico y el
resto tiene proteinuria subnefrótica (<3,5 g/24 h). La hematuria
microscópica es común (30% a 40%), pero la hematuria macroscópica y los
cilindros de glóbulos rojos son raros. Al momento de la presentación, la
mayoría de los pacientes no son hipertensos (<20 %) y la mayoría no tiene
insuficiencia renal (<20 %). En pacientes con síndrome nefrótico grave,
pueden surgir manifestaciones clínicas de hipercoagulabilidad (trombosis venosa
profunda, embolia pulmonar o trombosis de la vena renal).
En la biopsia renal, la lesión característica en el
microscopio óptico es un engrosamiento global, uniforme y difuso de la GBM en
ausencia de hipercelularidad mesangial o endocapilar significativa (fig. 63.5).

Figura 63. 5. Glomerulopatía membranosa.
El tratamiento con glucocorticoides solos es una
terapia insuficiente para la glomerulopatía membranosa. El tratamiento
recomendado es con una combinación de esteroides y agentes alquilantes como
ciclofosfamida o clorambucilo. Se desconoce la eficacia del micofenolato
mofetilo para el tratamiento de la MN. Rituximab, un anticuerpo monoclonal
anti-CD20, se ha encontrado eficaz en series de casos e informes de casos, y
actualmente se está realizando un ensayo controlado aleatorizado (el estudio
MENTOR [Membranous Nephropathy Trial of Rituximab] dirigido por Fervenza et
al.). Un estudio piloto de Waldman et al. del NIH ha informado resultados
alentadores usando una combinación de ciclosporina y rituximab (Kidney Int Rep,
2016). Una serie de casos reciente de 15 pacientes (Cortazar et al., 2017) con
una mediana de seguimiento de 37 meses apoya el uso de una terapia combinada
con rituximab, dosis bajas de ciclofosfamida oral y una disminución acelerada
de prednisona (revlimid ciclofosfamida prednisona [RCP ]). El cien por ciento
de los pacientes lograron una remisión parcial y el 93% de los pacientes
lograron una remisión completa en una mediana de tiempo de 2 y 13 meses. Los
autores citan la importancia de realizar estudios más amplios antes de sacar
conclusiones definitivas. El bloqueo de angiotensina en dosis altas con un
inhibidor de la enzima convertidora de angiotensina (ACE) o un bloqueador del
receptor de angiotensina (ARB) debe usarse para tratar la hipertensión y
controlar la proteinuria. La enfermedad puede recurrir en el trasplante renal,
pero esto es relativamente poco frecuente (10-15%).
El pronóstico de la MN depende en gran medida de la
cantidad de proteinuria. Los pacientes con proteinuria en rango nefrótico, más
de 6 g durante más de 6 meses, tienden a seguir un curso progresivo. Las
mujeres jóvenes con proteinuria moderada tienden a evolucionar muy bien; a los
machos mayores les va peor. Las remisiones completas o parciales espontáneas de
la proteinuria ocurren en aproximadamente el 40% de los pacientes, generalmente
dentro de los 3 a 5 años posteriores al diagnóstico. En general, 20 años
después del diagnóstico, alrededor de un tercio de los pacientes habrá
desarrollado ESRD, alrededor de un tercio estará en remisión y alrededor de un
tercio tendrá niveles variables de proteinuria persistente y función renal.
Entre los que experimentan remisión espontáneamente o con fármacos, alrededor
del 67 % permanece en remisión, mientras que el resto tiene recaídas
recurrentes sin progresión a insuficiencia renal (20 %) o progresión a
insuficiencia renal (13 %).
SÍNDROME NEFRÍTICO
La glomerulonefritis se define como la inflamación
aguda del compartimento glomerular. La nefritis resulta de la lesión de uno o
más de los tipos de células o estructuras que componen el glomérulo; células
endoteliales, epiteliales o mesangiales; o la membrana basal. La lesión se
puede clasificar en varios patrones patológicos diferentes, que se agrupan
ampliamente en tipos no proliferativos o proliferativos. La etiología de estos
diferentes tipos de nefritis puede ser de causas primarias, es decir,
intrínsecas al riñón, o de causas secundarias, que están asociadas a ciertas
infecciones (bacterianas, virales o parasitarias), fármacos, trastornos
sistémicos ( LES, vasculitis) o diabetes (Cuadro 63.2).

Cuadro 63. 2. Causas de nefritis.
Las pruebas serológicas para autoanticuerpos y la evaluación del patrón de hipocomplementemia suelen ser muy útiles en el estudio de los pacientes (tabla 63.1).

Tabla 63. 1. Hipocomplementemia en el estudio de
enfermedad glomerular
En última instancia, sin embargo, la biopsia renal
suele ser necesaria para hacer un diagnóstico definitivo.
El síndrome nefrítico se caracteriza por hematuria,
cilindros de glóbulos rojos, azotemia, proteinuria variable, edema e
hipertensión.
CAUSAS IMPORTANTES DEL SÍNDROME NEFRÍTICO
GLOMERULONEFRITIS PROLIFERATIVA DIFUSA AGUDA
(GLOMERULONEFRITIS POSTINFECCIOSA)
Este síndrome se observa con mayor frecuencia en niños
y con menos frecuencia en adultos. Los varones se ven afectados con mayor
frecuencia (proporción hombre:mujer [M:F] 2:1). La presentación es esporádica o
epidémica, de 1 a 3 semanas después de la infección con cepas nefritogénicas
del estreptococo beta-hemolítico del grupo A (cepas faríngeas 12, 2, 1 y 25)
que afectan la garganta o de 3 a 6 semanas después de una infección cutánea.
infección (pioderma cepas 49, 2, 42). El retraso en los síntomas renales
después de la infección de garganta o piel está relacionado con el período de
tiempo requerido para producir los anticuerpos que median la enfermedad renal.
La presentación clínica se caracteriza por un inicio
repentino de orina de color cola, hinchazón de la cara y los párpados,
proteinuria moderada e hipertensión.
Las pruebas de laboratorio incluyen títulos elevados
de antiestreptolisina O (ASLO) (en casos de infecciones de la piel, la
anti-ADNasa-B y la antihialuronidasa son más a menudo positivas) y disminución
de C3 y CH50. Los hallazgos positivos adicionales en PIGN incluyen velocidad de
sedimentación globular elevada, glóbulos rojos dismórficos en la orina y
cilindros de glóbulos rojos. La reducción de C3 rara vez persiste durante >8
semanas (un hallazgo importante para diferenciarlo de la glomerulonefritis
membranoproliferativa, que muestra una disminución persistente en los niveles
de complemento). En niños, con manejo sintomático, el 95% se recupera
clínicamente a los 2 meses del inicio y morfológicamente a los 3 años, aunque
algunos pueden progresar a la cronicidad.
Muchas otras infecciones pueden producir GNIP que se
asemejan a la GN posestreptocócica. Estos incluyen, entre otros, Mycoplasma
pneumoniae, citomegalovirus, Streptococcus pneumoniae, Neisseria meningitis,
Salmonella, toxoplasmosis, difteroides, especies de Propionibacter y
Staphylococcus aureus y albus.
La patología renal se caracteriza por una hipercelularidad
global de las células endoteliales y mesangiales en los glomérulos y un aumento
de la matriz mesangial. Hay abundante infiltración de células
polimorfonucleares dentro de los capilares glomerulares; esto a menudo se
denomina GN "exudativa". Pueden estar presentes medias lunas
ocasionales. La inmunofluorescencia muestra tres patrones de depósitos
inmunitarios: un patrón de cielo estrellado, que es el más típico, un patrón de
guirnalda y un patrón mesangial. Los depósitos son IgG y C3 con presencia variable
de IgM. La microscopía electrónica muestra “jorobas” subepiteliales y depósitos
mesangiales.
El tratamiento debe centrarse en eliminar la infección
estreptocócica con antibióticos y brindar terapia de apoyo hasta que ocurra la
resolución espontánea de la inflamación glomerular. Incluso si el paciente
presenta AKI, el pronóstico a corto plazo es bueno y >95% de los niños se
recuperarán del episodio inicial. La terapia inmunosupresora y la diálisis rara
vez son necesarias. Los adultos con PIGN tienen un 60 % de posibilidades de
recuperación. La proteinuria persistente es un signo de mal pronóstico.
La PIGN en los ancianos es causada con mayor
frecuencia por una infección estafilocócica y, a menudo, ocurre en un contexto
de diabetes. El alcoholismo y el uso de drogas por vía intravenosa también son
factores predisponentes.
NEFROPATÍA IGA O ENFERMEDAD DE BERGER
En 1968 Berger y Hinglais describieron un grupo de
pacientes con macrohematuria episódica, microhematuria persistente y
proteinuria moderada. Sus biopsias renales revelaron una lesión glomerular
proliferativa con depósito dominante de IgA en los glomérulos. La forma aguda
fue caracterizada por Volhard y Fahr como un episodio “sinfaringítico” (casi
siempre causado por S. aureus y albus) de hematuria, por lo general sin
azotemia, edema ni hipertensión.
La nefropatía por IgA (IgAN) tiene una prevalencia
estimada de 25 a 50 casos por cada 100 000 personas (considerada por algunos
como la forma más común de GN en todo el mundo). Es más común entre las
poblaciones asiáticas y nativas americanas y poco frecuente en los
afroamericanos. Todas las edades pueden verse afectadas; sin embargo, la IgAN
es más prominente en la segunda y tercera décadas (80% entre los 16 y los 35
años). Los hombres se ven más afectados que las mujeres. La predisposición
genética es probablemente cuestionable porque no hay asociaciones consistentes
con el antígeno leucocitario humano (HLA), excepto que HLA-B35 es más común en
pacientes franceses. También hay casos raros descritos de una forma familiar de
IgAN que se asocia con sordera. La púrpura de Henoch-Schönlein es una forma
sistémica de NIgA con vasculitis que se manifiesta como lesiones purpúricas
palpables, dolores abdominales y dolores articulares.
La hematuria macroscópica suele ser el primer síntoma
de presentación 24 a 48 horas después de una infección faríngea o
gastrointestinal, vacunación o ejercicio extenuante. Los casos asintomáticos se
presentan con hematuria microscópica durante un examen físico de rutina. Hay
una variedad de presentaciones que van desde hematuria macroscópica
intermitente hasta microhematuria persistente. Los síntomas constitucionales
(fiebre, dolores musculares, malestar general, fatiga y dolor en el
flanco/abdomen) a menudo acompañan a la nefritis.
No hay pruebas específicas aparte de una biopsia renal
que sean diagnósticas de IgAN.
Aunque no específicos, los niveles de IgA pueden estar
elevados en >50% de los pacientes. C3/C4 puede ser normal o incluso elevado
en algunos pacientes. El hallazgo anatomopatológico típico es una GN
mesangioproliferativa focal o difusa y depósitos inmunitarios de IgA mesangiales
difusos en la inmunofluorescencia (fig. 63.6), que se depositan en un patrón
granular difuso localizado principalmente dentro del mesangio.

Figura 63. 6. Nefropatía por IgA. (A) Microscopía
óptica. (B) Tinción mesangial IgA en inmunofluorescencia.
Existen clasificaciones patológicas, como la
clasificación de Haas, que clasifican la NIgA en cinco clases: (I) celularidad
mesangial mínima o nula; (II) lesiones de la GEFS; (III) GN proliferativa
focal; (IV) GN proliferativa difusa; y (V) >40% de glomeruloesclerosis
global y atrofia tubular. La clasificación de Oxford enfatiza características
histológicas pronósticas como hipercelularidad mesangial, hipercelularidad
endocapilar y esclerosis segmentaria.
No existe un tratamiento específico para la NIgA. Los
inhibidores de la ECA retardan la progresión del deterioro renal (algunos
estudios sugieren que esto ocurre más particularmente en pacientes con genotipo
DD del gen de la ECA). Se ha utilizado el tratamiento con esteroides y algunos
estudios sugieren eficacia. Sin embargo, el uso de esteroides sigue siendo
controvertido y puede ser más efectivo en pacientes IgA con síndrome nefrótico
y enfermedad de cambios mínimos. El tratamiento con aceite de pescado es
controvertido; un metaanálisis sugiere que los pacientes con alto riesgo de
progresión (sexo masculino, hipertensión, presencia de microhematuria,
presencia de proteinuria e insuficiencia renal) pueden beneficiarse. Una
combinación de ciclofosfamida oral, dipiridamol y warfarina en dosis bajas no
ha demostrado beneficios a largo plazo. De manera similar, la ciclosporina no
ha demostrado beneficio. Un reciente ensayo aleatorizado de Rauen et al. (2015)
que compararon la inmunosupresión con la atención de apoyo (bloqueo del sistema
renina-angiotensina) demostraron una tasa de remisión completa tres veces
mayor, una tasa más alta de resolución de la hematuria y una reducción de la
proteinuria entre los pacientes tratados con inmunosupresión en comparación con
los asignados a cuidado de apoyo solo. Sin embargo, no hubo cambios en la
disminución de la TFGe entre los dos grupos, y los pacientes tratados con
inmunosupresión tuvieron una tasa más alta de infecciones graves y otros
eventos adversos.
Adaptando las recomendaciones de una revisión reciente
de Pozzi (2016), se puede considerar el siguiente enfoque en pacientes con
NIgA:
1. En pacientes con hematuria macro-microscópica y
proteinuria < 0,3 g/d, seguimiento anual
2. En pacientes con proteinuria entre 0,3 y 0,9 g/d,
ACE-I y/o ARB, con titulación de los fármacos para reducir al máximo la
proteinuria según la tolerancia
3. En pacientes con proteinuria >1 g/día,
hipertensión y TFG >30 ml/min/1,73 m2, curso de 6 meses de corticosteroides
además de ACE-I o ARB
4. En pacientes con GFR <30 ml/min, ACE-I/ARB con
corticosteroides considerados para pacientes con proteinuria persistentemente
alta o creciente
5. En pacientes con IgAN rápidamente progresiva o
lesiones vasculíticas en la biopsia renal, se pueden considerar
inmunosupresores (ciclofosfamida y azatioprina).
El pronóstico general es variable y depende de la
clase histológica al inicio del estudio. Entre todos los que presentan IgAN,
del 20 % al 50 % de los pacientes eventualmente desarrollarán ESRD dentro de
los 20 años. Los marcadores de peor pronóstico son hipertensión persistente,
proteinuria, síndrome nefrótico, microhematuria persistente, edad avanzada y
sexo masculino (presente en la presentación inicial).
GLOMERULONEFRITIS RÁPIDAMENTE PROGRESIVA O EN SEMILUNAS
La presentación es una nefritis aguda de inicio abrupto
caracterizada por una rápida disminución de la función renal (una duplicación
de la creatinina sérica o una reducción del 50% en la TFG dentro de un período
de 3 meses). Por lo general, los pacientes progresan rápidamente a
insuficiencia renal en cuestión de semanas. Un diagnóstico certero y urgente es
fundamental en estos pacientes. La biopsia renal y el control de serologías
(anticuerpos anticitoplasma de neutrófilos [ANCA], anticuerpos anti-GBM,
anticuerpos antinucleares, anticuerpos anti-dsDNA y complementos) son
fundamentales. La biopsia renal revela una GN en media luna.
CUATRO CLASES DE GLOMERULONEFRITIS CRESCENTIC BASADAS
EN MECANISMOS PATOGENÉTICOS
Cuatro clases de GN semilunar son el tipo 1 (anti-GBM
positivo), el tipo 2 (enfermedad por inmunocomplejos; anti-GBM y ANCA
negativos), el tipo 3 (pauciinmune; ANCA positivo) y el tipo 4 (enfermedad con
doble anticuerpo positivo). , que tiene características de ambos tipos 1 y 3).
NEFRITIS DE LA MEMBRANA BASAL ANTIGLOMERULAR (TIPO 1)
La nefritis anti-GBM se presenta con insuficiencia
renal rápidamente progresiva con hematuria y cilindros de glóbulos rojos. Una
presentación que incluye disnea y hemoptisis (es decir, hemorragia pulmonar) se
denomina síndrome de Goodpasture. Hay una distribución bimodal con el primer
pico a los 30 años y el segundo pico a los 60 años. En 20% a 60% de los
pacientes, hay un pródromo de una infección del tracto respiratorio superior
que precede a la enfermedad. Los pacientes suelen tener proteinuria
subnefrótica (< 3 g/24 h), hematuria, sedimento urinario nefrítico y anemia
microcítica hipocrómica del tipo ferropénico. La hipertensión es poco común
(20% de los pacientes). Los factores de riesgo para la nefritis anti-GBM
incluyen antecedentes de exposición a hidrocarburos, tabaquismo, polvo
metálico, d-penicilamina, cocaína e infecciones por influenza A2.
La nefritis anti-GBM es causada por la presencia de
autoanticuerpos anti-GBM que reaccionan con el dominio no colagenoso de la
cadena alfa-3 del colágeno tipo IV. También se pueden observar autoanticuerpos
contra el dominio 1 no colagenoso de la cadena alfa-5 del colágeno tipo IV.
Este colágeno se expresa predominantemente en la membrana basal capilar
alveolar glomerular y pulmonar. Los autoanticuerpos anti-GBM se unen al antígeno
después de una lesión crónica de GBM. Esta reacción induce la activación del
complemento y el reclutamiento de leucocitos, lo que resulta en GN
proliferativa segmentaria necrosante, ruptura de las paredes capilares y,
finalmente, formación de medias lunas.
Las anomalías patológicas son llamativas (fig. 63.7).
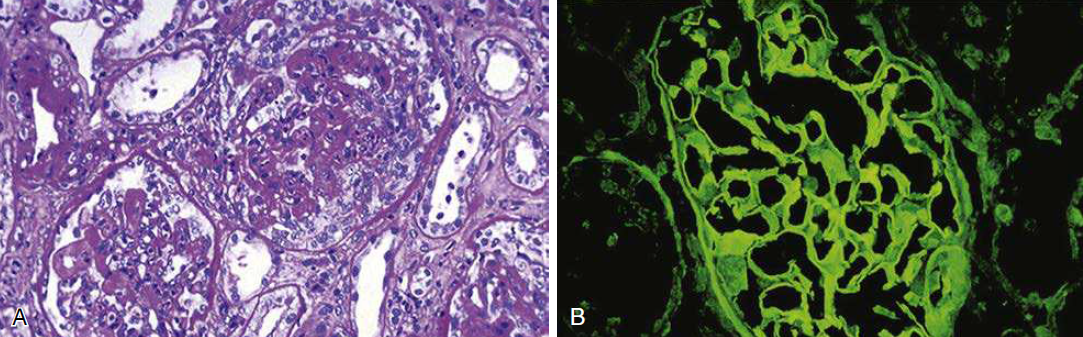
Figura 63. 7. Nefritis antimembrana basal glomerular.
(A) Microscopía óptica. (B) Tinción lineal de IgG en inmunofluorescencia.
El microscopio óptico muestra una GN en forma de media
luna difusa con depósitos lineales de IgG a lo largo de la GBM en la
inmunofluorescencia (se observa un patrón de inmunofluorescencia similar en las
biopsias de la membrana basal alveolar). La mayoría de las veces es IgG1. Se ha
informado IgG4, pero en tales casos, es una afectación débil de los riñones.
El curso clínico es de rápido deterioro de la función
renal (días a semanas), frecuentemente con necesidad de diálisis. Una
radiografía de tórax que muestre sombreado intersticial basilar e hiliar
bilateral anormal indicaría claramente una hemorragia pulmonar asociada y un
diagnóstico de síndrome de Goodpasture. El ensayo inmunoabsorbente ligado a
enzimas (ELISA) anti-GBM se puede utilizar para detectar anticuerpos anti-GBM
circulantes; el ensayo es específico para el dominio NCI de la cadena alfa-3
del colágeno tipo IV. Este anticuerpo se detecta en más del 90% de las personas
con nefritis anti-GBM. El nivel de creatinina plasmática suele ser un buen
indicador del grado de progresión. El estándar de oro para el diagnóstico es la
biopsia renal.
Sin tratamiento, los pacientes con nefritis anti-GBM
y/o síndrome de Goodpasture tienen un pronóstico muy pobre. La mayoría de los
pacientes mueren por complicaciones renales o pulmonares graves. La ESRD no es
infrecuente en pacientes que son oligúricos, anúricos o presentan una
creatinina sérica de >5 a 7 mg/dL. El objetivo de la terapia es suprimir la
formación de nuevos anticuerpos y eliminar los anticuerpos preexistentes. Esta
estrategia ha reducido notablemente la mortalidad (<10% a 1 año). El
tratamiento consiste en plasmaféresis con plasmaféresis durante 2 semanas en
días consecutivos, 1 mg/kg de prednisona (después de un pulso inicial de
metilprednisona de 1 g/día durante 3 días consecutivos) y 2 a 3 mg/kg de
ciclofosfamida por vía oral una vez al día. Los pacientes con glomerulonefritis
rápidamente progresiva (RPGN) tienen un peor pronóstico. En pacientes que
presentan hemorragia pulmonar (síndrome de Goodpasture), se recomienda la
plasmaféresis de urgencia. El punto final del tratamiento es cuando los
anticuerpos anti-GBM son indetectables en la sangre.
NEFRITIS LÚPICA (EJEMPLO DE TIPO 2)
La afectación renal se observa con frecuencia en
pacientes con LES. La prevalencia global de LES es de 12 a 64 casos por
100.000. La nefritis lúpica (NL) afecta predominantemente a mujeres en edad
fértil (relación M:F = 1:9); El 85% de los pacientes tendrán una edad <55
años. El SLE parece ser más común y tener una afectación renal más grave en la
población afroamericana. Entre el 40% y el 85% de los pacientes con LES tendrán
complicaciones que involucran el riñón (NL), pero el espectro de la enfermedad
renal (clase histológica) es muy variable. No existe un patrón genético
claramente definido para la predisposición al LES. Sin embargo, un porcentaje
importante de familiares de pacientes con LES desarrollan la enfermedad
(5-12%). Además, existe evidencia de un papel predisponente de los factores
hormonales, incluido el fuerte predominio de mujeres en edad fértil y la mayor
incidencia de LES en mujeres posmenopáusicas que toman estrógenos. Otros
factores predisponentes incluyen la exposición a la luz solar, la radiación
ultravioleta, los medicamentos y la exposición viral/bacteriana.
El LES puede afectar a cualquier sistema orgánico (ver
cuadro 63.3).

Cuadro 63. 3. Afectación de órganos en el lupus
eritematoso sistémico
La afectación renal a menudo se desarrolla al mismo
tiempo o poco después del inicio del LES y puede seguir un curso prolongado con
períodos de remisión y exacerbaciones. Un subgrupo raro especial de pacientes
("NL silencioso") no tiene hallazgos clínicos de afectación renal,
pero presentará NL proliferativa en la biopsia. La transformación de una clase
a otra es relativamente frecuente.
El diagnóstico de LES se basa tanto en criterios
clínicos como de laboratorio. La American Rheumatism Association ha
desarrollado criterios bajo los cuales se requieren 4 de los 11 hallazgos para
una sensibilidad y especificidad del 96% para diagnosticar SLE (Tabla 63.2).

Tabla 63. 2. Criterios de la American Rheumatism
Association para el diagnóstico de lupus eritematoso sistémico.
Los criterios abarcan las siguientes características
clínicas y de laboratorio: erupción malar; lupus discoide; enfermedad dérmica;
fotosensibilidad; ulceraciones orales o nasales; artritis no deformante; serositis,
incluyendo pleuritis y pericarditis; enfermedad del sistema nervioso central,
como convulsiones o psicosis; compromiso hematológico manifestado por la
presencia de anemia, leucopenia, linfopenia o trombocitopenia; marcadores
inmunológicos de enfermedad manifestados por una prueba de banda de lupus
positiva, una prueba de anticuerpos anti-ADN o anti-Sm positiva, una prueba de
laboratorio de investigación de enfermedades venéreas con resultado positivo
falso o una reacción de anticuerpos antinucleares (ANA) positiva; y afectación
renal, definida como proteinuria persistente superior a 500 mg diarios (3+ en
la tira reactiva), o la presencia de cilindros celulares, consistentes en
eritrocitos, hemoglobina, granulares, tubulares o mixtos.
La biopsia renal es una parte importante del
tratamiento de la NL y debe considerarse en todos los pacientes tanto para el
diagnóstico como para el tratamiento (fig. 63.8).

Figura 63. 8. Nefritis lúpica clase IV. Microscopía de
luz.
La Clasificación de la Sociedad Internacional de
Nefrología y Patología Renal (ISN/RPS) se utiliza para clasificar la NL. Esta
clasiicación divide a LN en varias clases (Tabla 63.3).
Tabla 63. 3. Clasificación de la nefritis lúpica de la
Sociedad Internacional de Nefrología y Patología Renal (Clasificación ISN/RPS)
La clase más común (en casi el 50% de los pacientes)
es la nefritis lúpica proliferativa difusa (NL clase IV). La enfermedad de
clase IV generalmente se presenta con todas o la mayoría de las características
del síndrome nefrítico agudo o RPGN.
La presencia de actividad y la cronicidad de la
enfermedad se incorporan a la clasificación ISN/RPS LN. Las características de
actividad incluyen hipercelularidad endocapilar, lesiones en asa de alambre,
necrosis fibrinoide, semilunas celulares e inflamación intersticial. La
cronicidad incluye esclerosis glomerular, atrofia tubular, fibrosis
intersticial y semilunas fibrosas. La enfermedad de clase II conlleva un buen
pronóstico y es posible que no esté indicado el tratamiento. Es difícil predecir
el curso y pronóstico de los pacientes clase III debido a su curso variado. Los
esteroides y la terapia citotóxica suelen ser necesarios para tratar la NL de
clase III. Los pacientes con clase IV requieren esteroides y terapia
citotóxica. El estándar de atención en muchos hospitales para las formas graves
de la enfermedad de clase IV son tres dosis consecutivas de metilprednisona en
pulsos (1 g por vía intravenosa cada día), seguidas de una dosis alta de
prednisona que comienza con 1 mg/kg/d y se reduce durante 3 meses hasta
aproximadamente 10 a 30 mg/día. Se recomienda la terapia citotóxica adyuvante.
Existe debate sobre si debe administrarse ciclofosfamida en pulsos por vía oral
o intravenosa. En muchos centros, ahora se prefiere el ácido micofenólico (MMF)
a la ciclofosfamida debido a su eficacia similar pero al perfil de riesgo
adverso más bajo. También se prefiere el MMF a la azatioprina. Sin embargo,
algunos centros continúan usando la ciclofosfamida en dosis completa o en la
mitad de la dosis (este último régimen se denomina protocolo Euro-Lupus).
VASCULITIS ASOCIADA A ANTICUERPOS ANTINEUTRÓFILOS
CITOPLÁSMICOS (EJEMPLO DE TIPO 3): GRANULOMATOSIS DE WEGENER (RENOMBRADA COMO
GRANULOMATOSIS CON POLIANGITIS)
La vasculitis puede afectar los vasos sanguíneos
grandes, medianos o pequeños. La vasculitis de vasos pequeños puede
clasificarse adicionalmente como vasculitis asociada o no asociada con ANCA. La
vasculitis de vasos pequeños asociada con ANCA incluye poliangeítis
microscópica, granulomatosis con poliangeítis, síndrome de Churg-Strauss
(también conocido como granulomatosis eosinofílica con poliangeítis) y
vasculitis inducida por fármacos.
La granulomatosis con poliangeítis puede ocurrir a
cualquier edad de la vida; la incidencia máxima es en la cuarta a sexta década
de la vida. La granulomatosis con poliangeítis se asocia con títulos elevados
de ANCA. Los ANCA están dirigidos contra antígenos presentes dentro de los
gránulos primarios de neutrófilos y monocitos. Sigue siendo controvertido si
los ANCA son la causa o un epifenómeno.
La presentación clínica de la granulomatosis con
poliangeítis es bastante variable y va desde una presentación subclínica con
afectación progresiva de las vías respiratorias y hallazgos renales leves hasta
una presentación más fulminante con GN aguda o GNRP.
La granulomatosis con poliangeítis afecta
predominantemente a las vías respiratorias, pero no es infrecuente la
afectación vasculítica multisistémica. La afectación de las vías respiratorias
superiores incluye sinusitis, tinnitus y pérdida de la audición con secreción
ótica y dolor. Los síntomas del tracto respiratorio inferior incluyen tos con
disnea que progresa a hemoptisis y hemorragia alveolar.
La enfermedad multisistémica puede incluir piel (p.
ej., pápulas, púrpura), articulaciones (artralgias, artritis), ojos
(conjuntivitis, epiescleritis), sistema nervioso, hígado, tiroides, vesícula
biliar y corazón. La presentación renal varía desde una RPGN e insuficiencia
renal hasta una disminución más gradual de la TFG con proteinuria de rango no
nefrótico, hematuria y cilindros de glóbulos rojos.
La anormalidad serológica clave es la presencia de
ANCA detectados en inmunofluorescencia indirecta o ELISA. Hay dos tipos
principales de anticuerpos definidos por su patrón de tinción en la
inmunofluorescencia indirecta: citoplasmáticos o C-ANCA y perinucleares o
P-ANCA. El antígeno proteinasa-3-ANCA produce un patrón de tinción
citoplasmático (C-ANCA), mientras que la mieloperoxidasa-ANCA produce un patrón
de tinción perinuclear (P-ANCA). Entre el 88% y el 96% de los pacientes con
granulomatosis con poliangeítis son ANCA positivos, más comúnmente C-ANCA, pero
también se observa P-ANCA. La patología típica en la biopsia renal es una
glomerulonefritis necrotizante y semilunar segmentaria focal. La vasculitis
puede afectar las arterias, venas y capilares renales de tamaño pequeño y
mediano. La inmunofluorescencia muestra un patrón inmunitario pobre (es decir,
con un depósito mínimo de inmunoglobulina).
Los pacientes no tratados tienen una tasa de
supervivencia de 1 año de 20% a 50%. Recomendó que el tratamiento inicial sea
metilprednisona (500-1000 mg por vía intravenosa al día durante 3 días
consecutivos) seguido de dosis altas de prednisona oral (1 mg/kg/día) y
ciclofosfamida oral (2 mg/kg/día, sin exceder los 200 mg/día). d). La
ciclofosfamida intravenosa en pulsos es un régimen alternativo aceptable, pero
puede asociarse con una mayor tasa de recaída, especialmente si la dosis total
acumulada es menor que la utilizada en la terapia oral continua. La remisión
con corticosteroides y ciclofosfamida ocurre en 85% a 95% de los pacientes. La
ciclofosfamida intravenosa con plasmaféresis puede ser mejor en pacientes con
hemorragia pulmonar o en los que están gravemente enfermos. Sin embargo, no
todos los pacientes tratados con ciclofosfamida responden, y hasta la mitad de
todos los pacientes tratados con ciclofosfamida recaen dentro de los 5 años.
En un análisis post hoc de rituximab para el ensayo de
vasculitis asociada a ANCA, los pacientes con vasculitis asociada a ANCA y
afectación renal respondieron de manera similar a la inducción de la remisión
con rituximab más glucocorticoides en comparación con
ciclofosfamida/azatioprina más glucocorticoides durante 18 meses de
seguimiento. Además, los eventos adversos son similares. Una revisión Cochrane
(Walters et al., 2015) que examinó la efectividad de varios tratamientos para
la vasculitis asociada a ANCA con afectación renal llegó a las siguientes
conclusiones:
1. La plasmaféresis fue eficaz en pacientes con LRA
grave secundaria a vasculitis.
2. La ciclofosfamida en pulsos da como resultado un
mayor riesgo de recaída en comparación con el uso oral continuo pero con una
dosis total reducida.
3. La ciclofosfamida es el tratamiento de inducción
estándar, pero el rituximab y el MMF también fueron efectivos. La azatioprina,
el metotrexato y la leflunomida son eficaces como terapia de mantenimiento.
FUENTE:
The Brigham Intensive
Review of Internal Medicine. (2022)
Ajay K. Singh, MBBS, FRCP, MBA
Joseph Loscalzo, MD, PhD









