Una mujer de 30 años fue evaluada en consultorio
externo de neurooftalmología de un hospital debido a la pérdida de visión
monocular.
Dos meses antes de la presentación actual, durante el
tercer trimestre del primer embarazo de la paciente, desarrolló dolor de cabeza
y disminución de la visión en el ojo derecho. El paciente pensó que el dolor de
cabeza se sentía como migrañas anteriores, que habían sido poco frecuentes. Sin
embargo, la disminución de la visión no había sido una característica típica de
sus migrañas. Los colores aparecían tenues y la percepción de la luz disminuía.
Había dolor en el ojo derecho pero no dolor con el movimiento del ojo.
Cinco días después, fue evaluada por un optometrista.
Informó que solo podía ver formas y movimientos. En una prueba de campo visual,
hubo una respuesta normal en el ojo izquierdo pero ninguna respuesta en el ojo
derecho. Se le indicó que buscara evaluación en un departamento de emergencias
de otro hospital.
En el servicio de urgencias, la resonancia magnética
nuclear (RMN) de la cabeza, que se realizó sin la administración de material de
contraste intravenoso debido al embarazo, reveló un marcado agrandamiento del
nervio óptico derecho intraorbitario. El nervio apareció isointenso en
comparación con los músculos extraoculares en las secuencias potenciadas en T1 pero
apareció brillante en la secuencia potenciada en T2; los globos parecían
normales y los músculos extraoculares no estaban agrandados. Los resultados de
la venografía por resonancia magnética de la cabeza fueron normales.
Ingresó en el hospital y se inició tratamiento con
metilprednisolona intravenosa por un diagnóstico presunto de neuritis óptica.
Durante la hospitalización, su capacidad para ver los colores mejoró levemente,
pero su visión no mejoró; sólo podía ver movimientos de manos. El quinto día de
hospital, fue dada de alta con instrucciones para programar citas de
seguimiento con un neurólogo y un optometrista.
Un mes después del alta del otro hospital y 2 semanas
antes de la presentación actual, la paciente fue evaluada nuevamente por su
optometrista. La visión del ojo derecho había empeorado; ya no podía ver sombras
ni movimientos, por lo que fue remitida para valoración por un
neurooftalmólogo.
En la presentación actual a la clínica de neurooftalmología,
no informó mejoría en la visión y dolor continuo en el ojo derecho. Tenía
antecedentes de enfermedad de Lyme, que había sido diagnosticada durante el
segundo trimestre del embarazo y había sido tratada con amoxicilina. Los medicamentos
incluían vitaminas prenatales y sulfato ferroso. La paciente vivía con su
esposo en Nueva Inglaterra y trabajaba en el cuidado de la salud. Nunca había
fumado tabaco y no bebía alcohol ni consumía drogas ilícitas. Un hermano tenía
antecedentes de epilepsia infantil y su madre tenía degeneración macular.
En la exploración, la agudeza visual era de 20/15 en
el ojo izquierdo y no había percepción de luz en el ojo derecho. Las pupilas
eran simétricas y reactivas a la luz, aunque la respuesta en la pupila derecha
era lenta; un defecto pupilar aferente estaba presente en el ojo derecho. En la
prueba de color de Ishihara, todas las placas se identificaron con el ojo
izquierdo. En una prueba de campo visual, hubo una respuesta normal en el ojo
izquierdo. La prueba de color de Ishihara y la prueba de campo visual no se
pudieron realizar en el ojo derecho debido a la mala visión. Los resultados de
un examen con lámpara de hendidura fueron normales, sin células identificadas en
la cámara anterior o vítreo. En un examen de fondo de ojo ( Figura 1A y 1B), el
disco óptico del ojo derecho estaba inflamado y con palidez temporal; había una
pequeña área de hipopigmentación cerca de la mácula en el ojo izquierdo. La
tomografía de coherencia óptica reveló un aumento del grosor de la capa de
fibras nerviosas de la retina y un adelgazamiento circunferencial del complejo
de células ganglionares del ojo derecho; no se observaron anomalías en el ojo
izquierdo.

Figura 1. Imágenes funduscópicas.
Se realizó un examen de fondo de ojo 8 semanas después
del inicio de los síntomas. En una fotografía del ojo derecho (Panel A), el
disco óptico muestra palidez en comparación con el disco óptico del ojo
izquierdo (Panel B), así como márgenes borrosos debido a la hinchazón
(asteriscos) y evidencia sutil de vasos anormales en la cara nasal (flecha). Se
realizó un examen de fondo de ojo de seguimiento 16 semanas después del inicio
de los síntomas. En una fotografía del ojo derecho (Panel C), el disco óptico muestra
mayor palidez, menor hinchazón y mayor prominencia de los vasos anormales en la
cara nasal, un hallazgo compatible con vasos de derivación optociliar.
El nivel de creatinina en sangre fue de 0,56 mg por
decilitro (rango de referencia, 0,60 a 1,50 mg por decilitro), y el nivel de
fosfatasa alcalina en sangre fue de 147 U por litro (rango de referencia, 30 a
100 ). Los resultados de otras pruebas de función renal y hepática fueron
normales, al igual que los niveles de electrolitos en sangre, el hemograma
completo y los resultados de las pruebas de coagulación. El nivel de proteína C
reactiva en sangre fue de 15 mg por litro (valor de referencia, <8) y la
velocidad de sedimentación globular de 34 mm por hora (rango de referencia, 0 a
20). Un análisis de sangre para anticuerpos antinucleares fue positivo a un
título de 1:40 con un patrón moteado. Las pruebas de anticuerpos contra la
acuaporina-4 y la glicoproteína de oligodendrocitos de mielina (MOG) fueron
negativas. Se consideró el diagnóstico de neuritis óptica atípica.
Seis días después del parto del bebé, la paciente fue
admitida en este hospital y se obtuvieron estudios adicionales. Se realizó
punción lumbar. En el análisis del líquido cefalorraquídeo (LCR), no había
células nucleadas y los niveles de proteína, glucosa, albúmina e IgG eran
normales. No hubo bandas oligoclonales después de la concentración del LCR. La
prueba del LCR fue negativa para anticuerpos contra la sífilis y para
anticuerpos contra la acuaporina-4.
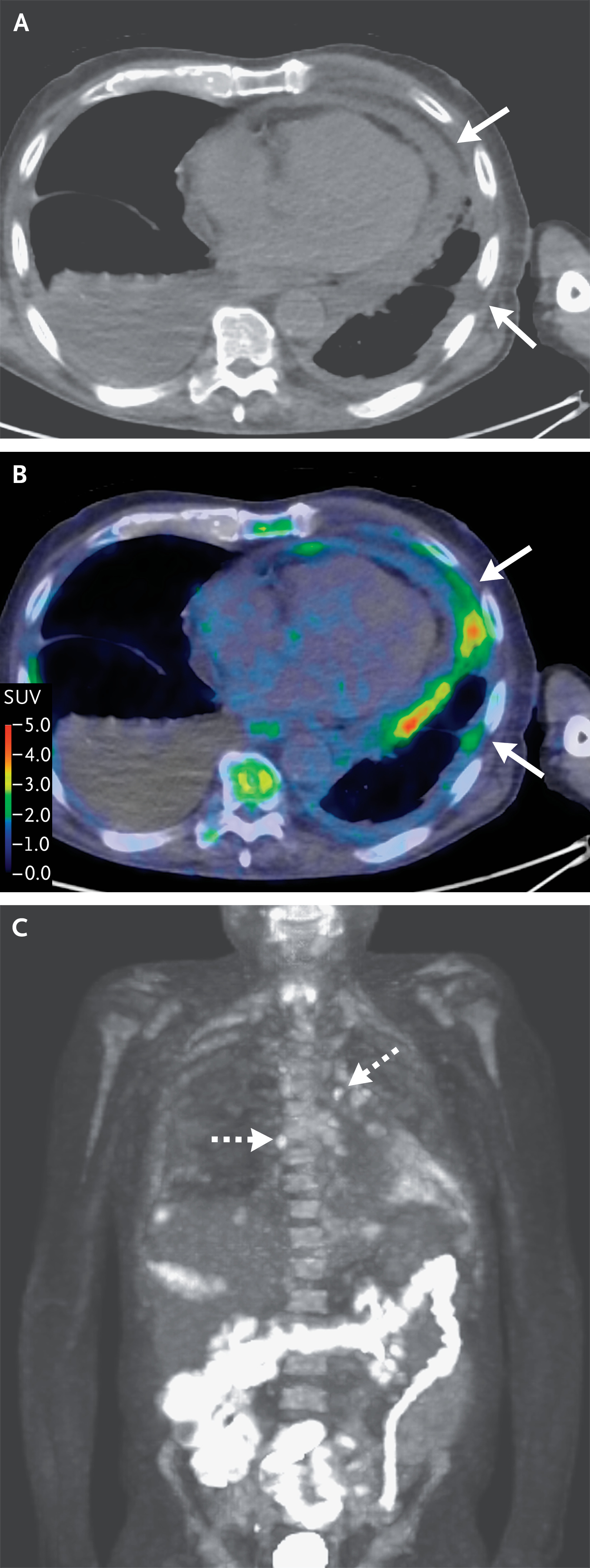
La resonancia magnética de la cabeza ( Figura 2 ),
realizada antes y después de la administración de material de contraste
intravenoso, reveló un agrandamiento fusiforme prominente de un segmento del
nervio óptico derecho intraorbitario con realce asociado, así como
hiperintensidad asociada en la secuencia de recuperación de inversión de tau
corta ponderada en T2 (STIR). No hubo evidencia de edema orbitario asociado u
otras anomalías orbitarias. Había un foco punteado hiperintenso dentro de la
sustancia blanca subcortical parietal izquierda en la secuencia de recuperación
de inversión atenuada por líquido potenciada en T2, sin realce asociado ni
difusión restringida.
Figura 2. Resonancia magnética de la cabeza.
Al ingreso en este hospital, aproximadamente 9 semanas
después del inicio de los síntomas, se realizó una resonancia magnética de la
cabeza antes y después de la administración de material de contraste
intravenoso. Las secuencias ponderadas en T1 realzadas con contraste coronal y
axial (Paneles A y B, respectivamente) muestran un agrandamiento fusiforme
prominente de un segmento del nervio óptico derecho intraorbitario con realce
homogéneo aumentado anormal (flechas). Una secuencia de recuperación de
inversión de tau corta ponderada en T2 coronal (STIR) (Panel C) muestra
hiperintensidad difusa que afecta a este segmento del nervio óptico derecho
(flecha) sin edema orbitario adyacente. Aproximadamente 2 meses después, se
realizó una resonancia magnética de seguimiento de la cabeza antes y después de
la administración de material de contraste intravenoso. Secuencias ponderadas
en T1 realzadas con contraste coronal y axial (Paneles D y E, respectivamente)
muestran un agrandamiento segmentario similar y un realce anormal del nervio
óptico derecho intraorbitario (flechas). Una secuencia STIR ponderada en T2
coronal (Panel F) muestra una hiperintensidad similar que afecta a este
segmento del nervio óptico derecho (flecha) sin cambios inflamatorios en la
grasa orbitaria.
Se administró tratamiento con metilprednisolona
intravenosa y plasmaféresis diaria. En el cuarto día de hospitalización, el
dolor en el ojo derecho se había resuelto y había percepción de luz en el ojo
derecho. Al quinto día de hospitalización se cambió el tratamiento con
metilprednisolona intravenosa a prednisona oral. En el noveno día de
hospitalización, se administró rituximab y fue dada de alta con prescripción
para un ciclo de prednisona de siete semanas de duración.
Durante las siguientes 3 semanas se administró
semanalmente tratamiento con rituximab. Dos semanas después de la última
infusión de rituximab, fue evaluada en la consulta de neurooftalmología. No
informó dolor en el ojo y solo dolor de cabeza ocasional, pero notó que la
visión en el ojo derecho había empeorado. A la exploración ojo derecho sin percepción
de luz, con defecto pupilar aferente. Los resultados de una prueba de campo
visual y la prueba de color de Ishihara no cambiaron con respecto a la
evaluación anterior en la clínica de neurooftalmología. En un examen de fondo
de ojo ( Figura 1C ), el disco óptico en el ojo derecho estaba hinchado y tenía
una palidez temporal más pronunciada; Los vasos de derivación optociliares
también estaban presentes recientemente. Se obtuvieron estudios de imagen
adicionales.
La RMN de seguimiento de la cabeza ( Figura 2 ) no
reveló ningún cambio radiológicamente significativo en el agrandamiento
segmentario del nervio óptico derecho intraorbitario, con realce persistente e
hiperintensidad en la secuencia STIR potenciada en T2. No se presentaron nuevas
anomalías orbitarias. Además, la lesión punteada en la sustancia blanca
subcortical parietal izquierda no se modificó.
Se realizó un procedimiento de diagnóstico.
Diagnóstico diferencial
Esta joven se presentó con pérdida de visión subaguda.
El primer paso en la evaluación de este paciente es localizar el problema en el
ojo, el nervio óptico o el cerebro. La paciente fue evaluado primero por un
oftalmólogo, quien examinó el ojo en busca de condiciones que pudieran conducir
a la pérdida de la visión monocular. 1 Cuando el examen del ojo no identificó
la causa de la pérdida de la visión, se evaluó a continuación en busca de
procesos que afectaran el nervio óptico o el cerebro. Esta evaluación debía
comenzar rápidamente para evitar más daños por una enfermedad potencialmente
tratable.
Las fibras del nervio óptico se extienden caudalmente
50 mm desde la retina hasta el quiasma óptico. Una lesión en cualquier parte
del nervio óptico puede causar pérdida de visión monocular. En el quiasma
óptico, que está encajado entre el hipotálamo del cerebro arriba y la glándula
pituitaria abajo, las vías ópticas comienzan a segmentarse en los campos
visuales derecho e izquierdo. La pérdida de visión en el campo visual derecho o
izquierdo se localiza en una lesión en el quiasma óptico, las vías ópticas, las
radiaciones ópticas en el cerebro o la corteza visual. En este paciente, el
examen reveló pérdida de visión monocular y no un defecto del campo visual en
ambos ojos. Centraré el diagnóstico diferencial en los procesos que afectan al
nervio óptico.
NEURITIS ÓPTICA
En una mujer joven, el diagnóstico diferencial de
enfermedades que afectan al nervio óptico comienza con trastornos
inflamatorios. Al considerar las posibles causas de la neuritis óptica
inflamatoria, es útil dividir el nervio óptico en tres segmentos: anterior,
intracanalicular y posterior.
El segmento anterior del nervio óptico tiene una
longitud de 25 mm y se extiende desde la retina hasta la entrada del cráneo.
Este segmento es comúnmente el blanco de la esclerosis múltiple. 2 Un total de
56% de los pacientes con esclerosis múltiple tienen afectación del nervio
óptico en algún momento del curso de la enfermedad. 3 Las lesiones suelen ser
focales (es decir, no largas) y se localizan dentro del propio nervio óptico.
La neuritis óptica es el evento inicial en el 20% de los pacientes que
presentan un nuevo diagnóstico de esclerosis múltiple; por esta razón, el
estudio de la neuritis óptica incluye resonancia magnética de la cabeza para
buscar las lesiones periventriculares que son típicas de la esclerosis
múltiple. 4En pacientes con esclerosis múltiple, la neuritis óptica
generalmente se cura sin tratamiento, pero la terapia a largo plazo para
prevenir ataques inflamatorios adicionales en el sistema nervioso central
reduce el riesgo de discapacidad a largo plazo. 5 En esta paciente, la
resonancia magnética de la cabeza no mostró lesiones que serían compatibles con
un diagnóstico de esclerosis múltiple. Además, no presentaba características
asociadas a un alto riesgo de este diagnóstico, como antecedentes familiares;
si tales características hubieran estado presentes, es posible que se haya
indicado una resonancia magnética anual de la cabeza. Mi sospecha general de
esclerosis múltiple en este paciente es baja.
El segmento anterior del nervio óptico también es un
objetivo de la neuritis óptica monofásica (también llamada neuritis óptica
idiopática). Los episodios a menudo ocurren después de que una infección o la
administración de una vacuna desencadenan una respuesta inmunológica. La
neuritis óptica monofásica generalmente se asocia con una alta probabilidad de
recuperación y un bajo riesgo de recaída. No existe una prueba de diagnóstico
para la neuritis óptica monofásica y, por lo general, se considera el
diagnóstico cuando se descartan causas alternativas. La esclerosis múltiple y
la neuritis óptica monofásica juntas representan el 95% de los casos de
neuritis óptica; cada uno de estos diagnósticos tiene la misma probabilidad de
ser la causa de la neuritis óptica. 4 Las causas menos comunes de neuritis
óptica incluyen la neurosarcoidosis, la enfermedad por anticuerpos MOG (MOGAD)
y el trastorno del espectro de la neuromielitis óptica (NMOSD).
La neurosarcoidosis es un proceso granulomatoso
sistémico, mientras que MOGAD y NMOSD son enfermedades autoinmunes específicas
de antígeno que se dirigen a MOG y al canal de agua acuaporina-4,
respectivamente. Los pacientes con neurosarcoidosis pueden presentar un curso
subagudo de neuritis óptica, que a menudo se desarrolla en el contexto de otras
neuropatías craneales, porque la inflamación tiende a extenderse a lo largo de
las meninges basales. 6 En pacientes con MOGAD, las lesiones que involucran el
nervio óptico tienden a ser largas, a veces se extienden a través de múltiples
segmentos del nervio óptico y causan inflamación meníngea (es decir,
perineuritis) que provoca dolor concomitante. 7En pacientes con NMOSD, los
segmentos intracanalicular y posterior del nervio óptico son blancos comunes de
la inflamación. El segmento intracanalicular del nervio óptico tiene solo 10 mm
de largo, pero la inflamación en esta parte del nervio es problemática debido
al espacio limitado para la inflamación dentro del canal óseo. Las lesiones de
NMOSD se encuentran entre las lesiones más destructivas asociadas con
enfermedades inflamatorias: la neuritis óptica debida a NMOSD causa ceguera en
el 60% de los pacientes a pesar del tratamiento con terapia inmunosupresora. 8
Tanto MOGAD como NMOSD son consideraciones en este
paciente debido a la pérdida severa de la visión y la inflamación extensa y el
realce del nervio óptico en la resonancia magnética. Para cada uno de estos
diagnósticos, las pruebas serológicas son muy específicas en el contexto de la
neuritis óptica; la sensibilidad de estos ensayos oscila entre el 60 y el 94 %.
9,10 En este paciente, los análisis de sangre para anticuerpos contra MOG y
acuaporina-4 fueron negativos. Aunque la enfermedad seronegativa es posible,
consideraré otras posibles explicaciones para la pérdida de visión de este
paciente.
INFECCIÓN
Las neuropatías ópticas infecciosas no son comunes en
los Estados Unidos. Las dos causas más probables en este paciente son
Bartonella henselae y Toxoplasma gondii . Cuando estos organismos afectan el
nervio óptico, también suele ocurrir inflamación de la retina, y este hallazgo
se identifica fácilmente en el fondo de ojo. En este paciente, la fundoscopia
mostró signos de congestión venosa del nervio óptico pero no evidencia de
infección. 11
TUMOR
La resonancia magnética de este paciente reveló una
lesión en el nervio óptico que tenía una apariencia de masa y no disminuyó de
tamaño a pesar de la administración de metilprednisolona intravenosa. La
presencia de neuritis óptica persistente o refractaria al tratamiento sugiere
NMOSD o neuropatía óptica sarcoide. Sin embargo, la presencia de una lesión de
masa persistente del nervio óptico puede ser indicativa de un tumor, más
comúnmente un glioma del nervio óptico o un meningioma de la vaina del nervio
óptico.
Los gliomas del nervio óptico surgen de los astrocitos
y forman masas de bajo grado similares a los astrocitomas pilocíticos de la
fosa inferior. 12 Más del 90% de los gliomas del nervio óptico ocurren en
personas menores de 20 años. 13 En este caso, la lesión observada en la
resonancia magnética tenía la apariencia clásica de un glioma del nervio
óptico, pero el paciente tenía 30 años de edad.
Los meningiomas de la vaina del nervio óptico surgen
del tejido meníngeo que rodea las fibras del nervio óptico y, por lo general,
comprometen la visión al comprimir el nervio óptico con el tiempo. 14 Debido a
que los meningiomas de la vaina del nervio óptico no suelen afectar el tejido
del nervio óptico, la resonancia magnética puede mostrar un signo
característico de “vía de tranvía”, un patrón causado por el realce simultáneo
del tejido tumoral y la falta de realce del nervio óptico. 15 Aunque el signo
de la vía del tranvía no se observó en la resonancia magnética en este paciente,
es más probable que ocurran meningiomas de la vaina del nervio óptico en
adultos que gliomas del nervio óptico. Para ambos tumores, el diagnóstico se
puede hacer mediante biopsia y el tratamiento consiste en resección quirúrgica
o radioterapia. La cirugía se asocia con un alto riesgo de ceguera en el ojo
afectado.
La biopsia de la vaina del nervio óptico rara vez se
realiza en el estudio de las neuropatías ópticas debido al riesgo de daño
permanente de la visión. Sin embargo, en este paciente, la mala respuesta a la
terapia inmunosupresora y el realce persistente y la apariencia de masa de la
lesión en la resonancia magnética hacen necesaria una biopsia para el
diagnóstico. Sospecho que la prueba diagnóstica en este paciente fue una
biopsia de la vaina del nervio óptico derecho.
IMPRESIÓN CLÍNICA
Esta mujer joven y embarazada se presentó con pérdida
de visión monocular dolorosa subaguda. La discromatopsia temprana progresó a
una disminución severa de la agudeza visual con un defecto pupilar aferente
relativo, características que sugieren una neuropatía óptica. Este diagnóstico
fue respaldado por una apariencia anormal del disco óptico, que tenía
inflamación, palidez y vasos de derivación optociliares. Una neuritis óptica
atípica fue el diagnóstico presuntivo inicial, pero la visión del paciente no
cambió después de la administración de inmunoterapia. En la resonancia
magnética repetida, el nervio óptico estaba demasiado agrandado y el realce
demasiado prolongado para un diagnóstico de neuritis óptica. Los resultados
normales de las pruebas serológicas y del análisis del LCR redujeron aún más la
probabilidad de inflamación, infección o infiltración por cáncer hematológico.
Por lo tanto,
La identificación de los vasos de derivación
optociliares proporcionó una pista importante en este caso. Los vasos de
derivación optociliar se forman cuando se ocluye el drenaje de la vena central
de la retina y la sangre fluye a través de las venas colaterales hacia las
venas coroideas peripapilares. 16 Los vasos de derivación optociliar ocurren
ocasionalmente en el contexto de otras condiciones, pero la asociación clásica
y pertinente es con meningiomas de la vaina del nervio óptico. Los meningiomas
pueden expresar receptores de progesterona y pueden crecer a un ritmo acelerado
durante el embarazo. 17 Sin embargo, la resonancia magnética no mostró la
apariencia radiológica típica de un meningioma de la vaina del nervio óptico;
de hecho, los hallazgos fueron más sugestivos de un glioma. 15Por lo tanto, se
recomendó una biopsia de la vaina del nervio óptico para distinguir entre estos
dos diagnósticos.
DIAGNOSTICO CLINICO
TUMOR DEL NERVIO ÓPTICO, YA SEA MENINGIOMA DE LA VAINA
DEL NERVIO ÓPTICO O GLIOMA DEL NERVIO ÓPTICO.
PROCEDIMIENTO DE DIAGNÓSTICO
Rara vez se realiza una biopsia de la vaina del nervio
óptico porque en aproximadamente el 2 % de los pacientes, el procedimiento
produce ceguera debido a una lesión vascular en el nervio óptico. Debido a que
este paciente tenía una pérdida de visión prolongada y palidez del nervio
óptico, no se esperaba una recuperación de la visión, por lo que la pérdida de
visión como complicación de la biopsia no se consideró un riesgo clínicamente
significativo.
Hay varios enfoques para realizar una biopsia de la
vaina del nervio óptico, pero la entrada a través del párpado superior medial
es la más común. Este abordaje sigue la ruta más corta hacia el nervio y no
requiere el desprendimiento de ningún músculo extraocular ni la extracción de
hueso. Además, la incisión se coloca en el pliegue del párpado, que tiene el
resultado cosméticamente más aceptable una vez que se completa la
cicatrización.
Figura 3. Fotografías Obtenidas durante Biopsia de la
Vaina del Nervio Óptico.
Las fotografías se obtuvieron a través de un microscopio
quirúrgico desde la vista del cirujano (invertida). El panel A muestra la
incisión a través del párpado superior medial derecho. El panel B muestra la
vaina del nervio óptico, que es gris y tiene el doble del tamaño de una vaina
normal del nervio óptico. El panel C muestra la vaina del nervio óptico
engrosada después de la incisión, con hipervascularización que ha provocado
sangrado. El panel D muestra el cierre de la incisión con suturas intestinales
al finalizar el procedimiento.
En este paciente, realizamos la biopsia después de
establecida la anestesia general. Se utilizó un microscopio quirúrgico ( Figura
3 ). Se realizó una incisión de 15 mm en el pliegue del párpado superior medial
derecho. Una vez incidido el tabique orbitario, se realizó disección roma hasta
visualizar el nervio. La vaina del nervio óptico tenía aproximadamente el doble
del diámetro de una vaina del nervio óptico normal y tenía un aspecto irregular
y muy vascularizado. Se tomó una muestra de una pequeña porción de la vaina del
nervio óptico. A pesar de la pérdida de visión del paciente, no se realizó una
sección completa del nervio.
DISCUSIÓN PATOLÓGICA
La tinción con hematoxilina y eosina de secciones de
la muestra de biopsia orbitaria ( Figura 4) mostró fragmentos de una neoplasia
eosinofílica mezclados con tejido fibroadiposo orbitario y un pequeño fragmento
de hueso. El tumor tenía una arquitectura claramente lobulada o anidada. Las
células neoplásicas parecían crecer en un patrón sincitial y tenían membranas
celulares indistintas, abundante citoplasma eosinofílico y núcleos redondos a
ovalados relativamente uniformes. Se identificaron pseudoinclusiones
intranucleares y no se detectaron calcificaciones laminadas concéntricas
(cuerpos de psammoma). No hubo características de un tumor maligno
(Organización Mundial de la Salud [OMS] grado 3) o meningioma atípico (OMS
grado 2), como aumento de la actividad mitótica, invasión glial, aumento de la
celularidad, crecimiento en forma de lámina, nucleolos prominentes o necrosis;
la ausencia de tales características era compatible con un tumor de bajo grado
(grado 1 de la OMS).Las células tumorales mostraron una tinción nuclear difusa
para el receptor de progesterona, que es una característica común de los
meningiomas de bajo grado, en particular los que afectan la vaina del nervio
óptico. 18,19 El índice de proliferación Ki-67, una medida pronóstica
importante para determinar el riesgo de recurrencia, incluso en pacientes con
tumores de bajo grado, fue muy bajo, del 0,7 % (8 células positivas de 1080
células contadas). 20

Figura 4. Muestra de biopsia de la vaina del nervio
óptico.
La tinción con hematoxilina y eosina muestra
fragmentos de un tumor eosinofílico mezclado con grasa orbitaria y hueso (Panel
A, flecha). A mayor aumento, la arquitectura del tumor es predominantemente
anidada o verticilada (Panel B, en un círculo); este patrón de crecimiento es
característico del meningioma meningotelial. Las células tienen bordes
celulares indistintos y núcleos monótonos, banales, redondos a ovalados con
pseudoinclusiones nucleares ocasionales (Panel C y recuadro, flechas). No hay
evidencia de características atípicas, como necrosis, figuras mitóticas o
invasión glial (Panel D). En la tinción inmunohistoquímica para el receptor de
progesterona, hay positividad nuclear difusa (Panel E, en marrón). En la
tinción inmunohistoquímica para Ki-67, el índice de proliferación es muy bajo,
con células positivas que constituyen menos del 1% de las células contadas
(Panel F, en rojo).
DIAGNÓSTICO PATOLÓGICO
MENINGIOMA, TIPO MENINGOTELIAL, GRADO 1 DE LA
ORGANIZACIÓN MUNDIAL DE LA SALUD.
DISCUSIÓN DEL MANEJO
Los meningiomas de la vaina del nervio óptico
generalmente se tratan con radioterapia. 21 El momento de la radioterapia para
un paciente con un meningioma de la vaina del nervio óptico se basa más
comúnmente en los hallazgos clínicos y de imagen, dado que la biopsia
generalmente no se intenta debido al riesgo de deterioro adicional de la
visión. Sin embargo, si se dispone de un diagnóstico histológico, puede ser útil
para confirmar la evaluación clínica y el plan de tratamiento.
El objetivo principal de la radioterapia es prevenir
un mayor crecimiento del tumor. El crecimiento tumoral descontrolado conduce a
la pérdida de la visión y proptosis gradual del ojo, lo que provoca
desfiguración y dolor y, finalmente, conduce a la enucleación. En la mayoría de
las series retrospectivas, la radioterapia ha resultado en un control duradero
del tumor en más del 90 % de los casos. 21,22 Más allá del control del tumor y
la prevención de cambios estéticos, los objetivos adicionales del tratamiento
son la retención de la visión y, a veces, la recuperación de la visión, que es
más probable que ocurra si el tratamiento se inicia dentro de las semanas o
meses posteriores al inicio de los síntomas. 22 Los pacientes con deterioro de
la visión de larga duración no suelen tener una recuperación de la visión.
En esta paciente, se administró radioterapia 5 días a
la semana durante un período de 6 semanas, con una dosis total administrada de
54 Gy en 30 fracciones. Para minimizar los efectos secundarios a largo plazo
asociados con la radioterapia, se limitó la irradiación colateral del globo
anterior, la glándula lagrimal, el tejido cerebral normal circundante, la
órbita contralateral, el quiasma óptico y la glándula pituitaria. La terapia de
protones se utilizó para administrar el nivel más bajo de radiación colateral a
los tejidos normales circundantes. La paciente respondió razonablemente bien al
tratamiento, aunque había esperado efectos secundarios, como dolor y presión
locales, ptosis, retraso en los movimientos oculares y fatiga. Un año después
de la radioterapia, se confirmó el control del tumor mediante imágenes y sus
síntomas fueron mucho menos graves, pero no había recuperado la visión.
PERSPECTIVA DEL PACIENTE
PACIENTE :Estaba en el tercer trimestre de mi embarazo
y tenía este dolor de cabeza palpitante mientras conducía. Cuando frotaba mi
ojo izquierdo y usaba mi ojo derecho para ver, todos los faros de los autos
parecían apagarse. Cuando se diagnosticó el tumor, habían pasado varios meses.
Nunca pensé que me caería por las grietas, pero casi lo hago. Pasar por el
tratamiento fue abrumador. Inicialmente me dijeron que no era un tumor y que
recuperaría la visión. De repente, tuve la experiencia de ser mamá primeriza,
someterme a un tratamiento, mantener la esperanza de recuperar la visión,
manejar el efecto en mi carrera y pasar la Navidad hospitalizada con un recién
nacido; estaba confusa durante ese período. Me tomó un año reflexionar sobre mi
experiencia. Lamento no haber presionado más para que me transfirieran a un
centro más grande cuando me hospitalizaron por primera vez. y me pregunto si mi
visión podría haberse recuperado si lo hubiera hecho. Cuando estaba en
tratamiento, mis proveedores dieron prioridad a mis objetivos posparto.
Aliviaba la culpa de no estar presente durante la licencia de maternidad. Si
saca algo de mi experiencia, espero que sea para tratar la pérdida de la visión
con urgencia porque podría significar la recuperación de la visión. Este
diagnóstico cambió mi vida, pero tengo suerte. Gracias a mi equipo. Me siento
afortunada de tenerlos a mi lado.
DIAGNOSTICO FINAL
MENINGIOMA, TIPO MENINGOTELIAL, GRADO 1 DE LA
ORGANIZACIÓN MUNDIAL DE LA SALUD.
Traducción de.
“A 30-Year-Old Woman with Decreased Vision and
Headache”
Michael Levy, M.D., Ph.D., Bart K. Chwalisz, M.D.,
Benjamin M. Kozak, M.D., Michael K. Yoon, M.D., Helen A. Shih, M.D., and Anna
M. Stagner, M.D.
NEJM
https://www.nejm.org/doi/full/10.1056/NEJMcpc2211355?query=featured_home
1. Galloway NR, Amoaku WMK, Gallo[1]way
PH, Browning AC. Common eye dis[1]eases
and their management. Cham,
Switzerland: Springer International Pub[1]lishing,
2016 (https://link.springer.com/
book/10.1007/978-3-319-32869-0).
2. Mealy MA, Whetstone A, Orman G,
Izbudak I, Calabresi PA, Levy M. Longitu[1]dinally
extensive optic neuritis as an MRI
biomarker distinguishes neuromyelitis
optica from multiple sclerosis. J Neurol
Sci 2015;355:59-63.
3. Frohman EM, Frohman TC, Zee DS,
McColl R, Galetta S. The neuro-ophthal[1]mology
of multiple sclerosis. Lancet Neu[1]rol
2005;4:111-21.
4. Optic Neuritis Study Group. Multiple
sclerosis risk after optic neuritis: final
optic neuritis treatment trial follow-up.
Arch Neurol 2008;65:727-32.
5. Gal RL, Vedula SS, Beck R. Cortico[1]steroids
for treating optic neuritis. Co[1]chrane
Database Syst Rev 2015;8:
CD001430.
6. Baughman RP, Weiss KL, Golnik KC.
Neuro-ophthalmic sarcoidosis. Eye Brain
2012;4:13-25.
7. Sechi E, Cacciaguerra L, Chen JJ, et al.
Myelin oligodendrocyte glycoprotein
antibody-associated disease (MOGAD):
a review of clinical and MRI features, di[1]agnosis,
and management. Front Neurol
2022;13:885218.
8. Levin MH, Bennett JL, Verkman AS.
Optic neuritis in neuromyelitis optica.
Prog Retin Eye Res 2013;36:159-71.
9. Gastaldi M, Scaranzin S, Jarius S,
et al. Cell-based assays for the detection
of MOG antibodies: a comparative study.
J Neurol 2020;267:3555-64.
10. Prain K, Woodhall M, Vincent A, et al.
AQP4 antibody assay sensitivity compari[1]son
in the era of the 2015 diagnostic cri[1]teria
for NMOSD. Front Neurol 2019;10:
1028.
11. Kahloun R, Abroug N, Ksiaa I, et al.
Infectious optic neuropathies: a clinical
update. Eye Brain 2015;7:59-81.
12. Nair AG, Pathak RS, Iyer VR, Gandhi
RA. Optic nerve glioma: an update. Int
Ophthalmol 2014;34:999-1005.
13. Huang M, Patel J, Patel BC. Optic
nerve glioma. Treasure Island, FL: Stat[1]Pearls,
2022 (http://www.ncbi.nlm.nih.gov/
books/NBK557878/).
14. Parker RT, Ovens CA, Fraser CL, Sa[1]marawickrama
C. Optic nerve sheath me[1]ningiomas:
prevalence, impact, and man[1]agement
strategies. Eye Brain 2018;10:
85-99.
15. Miller NR. Primary tumours of the
optic nerve and its sheath. Eye (Lond)
2004;18:1026-37.
16. Boschetti NV, Smith JL, Osher RH,
Gass JD, Norton EW. Fluorescein angiog[1]raphy
of optociliary shunt vessels. J Clin
Neuroophthalmol 1981;1:9-30.
17. Hortobágyi T, Bencze J, Murnyák B,
Kouhsari MC, Bognár L, Marko-Varga G.
Pathophysiology of meningioma growth
in pregnancy. Open Med (Wars) 2017;12:
195-200.
18. Thom M, Martinian L. Progesterone
receptors are expressed with higher fre[1]quency
by optic nerve sheath meningio[1]mas.
Clin Neuropathol 2002;21:5-8.
19. Maiuri F, Mariniello G, de Divitiis O,
et al. Progesterone receptor expression in
meningiomas: pathological and prognos[1]tic
implications. Front Oncol 2021;11:
611218.
20. Nowak-Choi K, Palmer JD, Casey J,
et al. Resected WHO grade I meningioma
and predictors of local control. J Neuro[1]oncol
2021;152:145-51.
21. de Melo LP, Arruda Viani G, de Paula
JS. Radiotherapy for the treatment of op[1]tic
nerve sheath meningioma: a system[1]atic
review and meta-analysis. Radiother
Oncol 2021;165:135-41.
22. Arvold ND, Lessell S, Bussiere M, et al.
Visual outcome and tumor control after
conformal radiotherapy for patients with
optic nerve sheath meningioma. Int J Ra[1]diat
Oncol Biol Phys 2009;75:1166-72