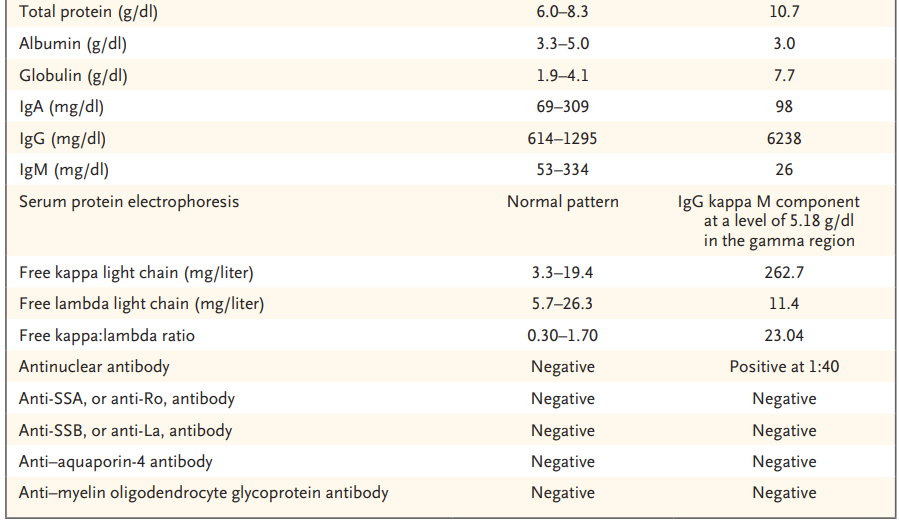
En este ejercicio clínico se presenta un caso que es discutido por un médico internista al que se le van proporcionando datos de la historia clínica en forma secuencial, y este analiza el cuadro a la luz de los nuevos elementos, de una manera análoga al proceso diagnóstico en la práctica real de la medicina
HISTORIA
Una mujer de 18 años previamente sana se presentó a su
médico de atención primaria con un historial de 2 meses de dolores de cabeza y
dolor medioescapular. El dolor medioescapular empeoraba en reposo,
particularmente por la noche, y disminuía con el ejercicio. Los síntomas
comenzaron después de una colisión de vehículos de motor a baja velocidad y 2
semanas después de haber recibido una vacuna de ARN mensajero contra la
enfermedad por coronavirus 2019 (Covid-19). La frecuencia cardíaca era de 85
latidos por minuto, la presión arterial de 114/65 mm Hg y la temperatura
corporal de 36,7 °C. Se observaron aproximadamente 12 úlceras orales en la
lengua y la mucosa bucal. Los exámenes musculoesqueléticos y cardiopulmonares
fueron normales. Dada la historia de 10 años del paciente de estomatitis
herpética intermitente, se prescribió un curso empírico de aciclovir. Se
recomendó naproxeno y fisioterapia para el dolor de espalda.
PONENTE
Aunque el dolor de espalda es un hallazgo
inespecífico, el dolor nocturno generalmente no se asocia con tensión muscular
y puede presagiar una causa más grave. Lo más probable es que las ulceraciones
orales en este paciente se deban a una infección por el virus del herpes simple
(VHS), pero también pueden ser una manifestación de una variedad de
enfermedades, que incluyen lupus eritematoso sistémico, artritis reactiva,
enfermedad de Behçet, enfermedad inflamatoria intestinal, enfermedad celíaca,
dermatología. (p. ej., enfermedad ampollosa autoinmune) e infección por el
virus de la inmunodeficiencia humana. La relevancia de la reciente vacunación
contra el Covid-19 es incierta, aunque ha habido informes anecdóticos de
ulceraciones orales después de la inmunización.
EVOLUCIÓN
El dolor de espalda de la paciente no disminuyó y
migró de la parte media de la espalda a la parte inferior de la espalda, y se
desarrollaron fiebres recurrentes, lo que motivó la derivación a una clínica de
reumatología, donde fue evaluada 2 meses después de la presentación inicial. No
informó dolor ocular, cambios en la visión, dolor abdominal, diarrea,
hematoquecia o antecedentes de trombosis venosa o arterial. Ella había sido
adoptada y no podía proporcionar antecedentes familiares. El examen no reveló
sinovitis o erupción. Mientras tanto, se había desarrollado una ulceración
vaginal que era similar a sus ulceraciones orales. Se pensó que la
espondiloartritis inflamatoria era una consideración diagnóstica importante y
se realizaron estudios de laboratorio, con resultados que incluyeron un
hemograma completo y un panel metabólico normales, una velocidad de
sedimentación globular (VSG) de 100 mm por hora (valor normal, ≤20), un nivel
de proteína C reactiva (PCR) sérica de 102 mg por litro (valor normal, ≤10),
prueba de detección de anticuerpos antinucleares negativa y un panel de
anticuerpos anticitoplasma de neutrófilos negativo. Los resultados de las
pruebas adicionales durante los siguientes meses incluyeron un nivel de
anticuerpos IgG contra el VHS tipo 1 (VHS-1) de 50,30 unidades (valor normal,
<0,91), positividad para anticuerpos contra la transglutaminasa tisular
(tTG) y anticuerpos contra la gliadina desamidada, y complemento sérico normal
niveles Las pruebas fueron negativas para HLA-B27 y HLA-B51 y para un panel de
15 anticuerpos asociados con miositis. La resonancia magnética nuclear (RMN) no
mostró evidencia de sacroilitis. La endoscopia superior, realizada debido al
nivel elevado de tTG, mostró, Helicobacter pylori, pero no mostró evidencias de
enfermedad celíaca. La infección por H pylori fue tratada, y se administró una
dieta libre de gluten por posible sensibilidad al gluten.
PONENTE
Los síntomas persistentes de la paciente y el nivel
marcadamente elevado de PCR y VSG sugieren un trastorno inflamatorio sistémico.
La ausencia de evidencia de sacroileítis argumenta en contra de la
espondiloartritis pero no la descarta. La enfermedad de Behçet, una vasculitis
multisistémica idiopática, puede causar ulceraciones orales y genitourinarias,
así como síntomas constitucionales. Contrariamente a la fuerte relación entre
HLA-B27 y la espondilitis anquilosante, HLA-B51 solo se asocia modestamente con
la enfermedad de Behçet, con valores predictivos positivos y negativos bajos.
El nivel elevado de anticuerpos HSV-1 IgG indica una infección previa por HSV,
pero no informa si las ulceraciones recientes se debieron a esta infección.
EVOLUCIÓN
Seis meses después del inicio de los síntomas y varias
semanas antes del seguimiento ambulatorio programado, la paciente se presentó
en el servicio de urgencias con un historial de 2 semanas de disnea de esfuerzo
progresiva y opresión en el pecho. La temperatura corporal de la paciente era
de 37,1°C, la frecuencia cardíaca de 107 latidos por minuto, la presión
arterial de 98/61 mm Hg, la frecuencia respiratoria de 15 por minuto y la
saturación de oxígeno del 99% mientras respiraba aire ambiente.
PONENTE
El diagnóstico diferencial de esta presentación aguda es
amplio e incluye embolia pulmonar y otras enfermedades pulmonares, anemia y
causas cardiovasculares, como enfermedad coronaria, miocarditis, pericarditis y
disfunción valvular. Con una historia de 2 semanas, es improbable un evento
coronario agudo, como disección o espasmo coronario, aunque es posible que
ocurra insuficiencia cardíaca varios días después del evento agudo. La
endocarditis puede manifestarse con un curso subagudo prolongado, seguido de un
deterioro más rápido cuando sobreviene una disfunción valvular. Sin embargo,
las fiebres recurrentes y el dolor de espalda junto con marcadores
inflamatorios elevados despiertan preocupación por la enfermedad aórtica
inflamatoria. La miocarditis o la pericarditis pueden ocurrir en pacientes con
un trastorno inflamatorio sistémico o una infección viral, lo que provoca
fiebre y finalmente insuficiencia cardíaca (en el último caso por
características fisiológicas constrictivas después de la pericarditis).
EVOLUCIÓN
En el examen físico destacaba un soplo diastólico
decreciente grado 4/6 que era más intenso en el borde esternal inferior
izquierdo y crepitantes en los campos inferior y medio de ambos pulmones. Un
soplo estaba presente en la fosa supraclavicular derecha. Los pulsos eran 2+ en
brazos y piernas. La paciente no tenía úlceras orales ni genitales ni exantema.
Los exámenes neurológico y musculoesquelético fueron normales. El hemograma
completo, el nivel de glucosa y los niveles de electrolitos fueron normales. El
nivel de creatinina sérica fue de 1,07 mg por decilitro, en comparación con un
nivel inicial de 0,69 mg por decilitro. El nivel de troponina I sérica fue de
0,05 ng por mililitro (valor normal, ≤0,04), el nivel de péptido natriurético
tipo B de 2484 pg por mililitro (valor normal, ≤101), el nivel de D-dímero 0,96
μg por mililitro (valor normal, ≤0,59), VSG 19 mm por hora y nivel de PCR 41,9
mg por litro. Una radiografía de tórax mostró cardiomegalia y aumento de las
marcas intersticiales, ambos nuevos en comparación con un estudio obtenido 8
meses antes en el momento de la colisión del vehículo de motor ( Figura 1 ).


Figura 1. Radiografías de tórax.
Comparada con una Rx de tórax normal en un estudio 8
meses antes (Panel A), la Rx anteroposterior actual (Panel B), mostró aumento
de la trama intersticial y una cardiomegalia nueva aun teniendo en cuenta la
diferencia de técnica entre ambas radiografías.
PONENTE
El examen es compatible con insuficiencia aórtica
debida a enfermedad de la válvula aórtica o características patológicas en la
aorta ascendente. Aunque una anomalía de larga data, como la válvula aórtica
bicúspide congénita, es una posible causa subyacente, el tamaño cardíaco normal
en la radiografía 8 meses antes sugiere un proceso más agudo.
La endocarditis es la causa más frecuente de
insuficiencia aórtica aguda. Sin embargo, el ensanchamiento del mediastino en
la radiografía de tórax despierta preocupación por un aneurisma o disección
aórtica. El agrandamiento de la silueta cardíaca puede reflejar la dilatación
del ventrículo izquierdo, pero también se debe considerar el derrame
pericárdico que complica la disección aórtica.
EVOLUCIÓN
Una angiografía por tomografía computarizada (TC)
reveló edema intersticial y alveolar sin evidencia de tromboembolismo. Se
observó que el contorno de la aorta toracoabdominal descendente era irregular
con estrechamiento proximal de las arterias mesentérica superior y tronco celíaco ( Figura 2 ). No hubo derrame
pericárdico.
Figura 2. Angiografía por TC.
Se ve un contorno aórtico irregular, con
estrechamiento asimétrico y concéntrico, así como una estenosis focal del 65%
del ostium de la arteria mesentérica superior (flecha).
PONENTE
Los hallazgos de la TC sugieren una aortitis
inflamatoria. La arteritis de Takayasu, una vasculitis de grandes vasos que
afecta con mayor frecuencia a mujeres menores de 40 años, es más probable. El
diagnóstico diferencial de la aortitis también incluye la enfermedad de Behçet,
aunque la afectación vascular en la enfermedad de Behçet afecta
predominantemente a hombres y rara vez involucra la raíz aórtica o la aorta
ascendente. Aunque las ulceraciones genitourinarias son consistentes con la
enfermedad de Behçet, la infección por HSV es otra explicación plausible de las
ulceraciones en este paciente. La paciente tampoco tiene evidencia de otros
hallazgos asociados con la enfermedad de Behçet, como uveítis, manifestaciones
neurológicas, trombosis o sinovitis, aunque la ausencia de estas condiciones no
descarta este diagnóstico. La aortitis infecciosa, especialmente la aortitis
sifilítica, es menos probable, dado que típicamente se manifiesta en adultos
mayores como una complicación de la sífilis terciaria, pero debe descartarse.
Se deben realizar imágenes de la válvula y la aorta.
EVOLUCIÓN
La ecocardiografía transtorácica, reveló dilatación
ventricular izquierda severa y función sistólica reducida (fracción de
eyección, 37%). Había una regurgitación aórtica severa dirigida anteriormente
de una válvula de tres valvas, con dilatación aneurismática del seno de Valsalva
izquierdo que resultaba en una mala coaptación de la cúspide coronaria
izquierda. Se calculó una fracción regurgitante del 56% con el uso de la
integral velocidad-tiempo Doppler. La ecografía dúplex arterial reveló
múltiples estenosis focales de hasta el 50% en la arteria subclavia derecha. La
prueba de punción con aguja fue negativa para patergia. La detección de
anticuerpos IgG e IgM de Treponema pallidum no fue reactiva.
PONENTE
Aunque no es definitiva, la ausencia de patergia
(definida como el desarrollo de una pápula o pústula estéril dentro de 1 o 2
días después de la inserción de la aguja) argumenta aún más en contra de la
enfermedad de Behçet. La dilatación del ventrículo izquierdo es un mecanismo
compensatorio desadaptativo que mantiene el gasto cardíaco normal a pesar del
reflujo diastólico sustancial y sugiere que la regurgitación aórtica no es
aguda. Se debe considerar la ecocardiografía transesofágica (ETE) o la TC con
control cardíaco con el uso de material de contraste para descartar la disección
y caracterizar la raíz aórtica antes de la intervención quirúrgica.
EVOLUCIÓN
La ETE confirmó insuficiencia aórtica grave por
aneurisma del seno de Valsalva izquierdo. La resonancia magnética cardíaca no
mostró cicatriz miocárdica y confirmó insuficiencia aórtica grave (fracción
regurgitante, 53%). La tomografía por emisión de positrones (PET) y la TC con
18 F-fluorodesoxiglucosa (FDG) revelaron inflamación aórtica activa difusa,
incluso en la raíz aórtica ( Figura 3 ).


Figura 3. Hallazgos FDG-PET y CT.
Las imágenes de TC con bajas dosis son co registradas
con imágenes de tomografía por emisión de positrones y 18F-fluorodeoxyglucose
(FDG), en la raíz aórtica y en el arco aórtico, indicado por las flechas en el
Panel A (vista axial), y Panel B (vista coronal). La captación aórtica de FDG
es sugestiva de inflamación.
PONENTE
La arteritis de Takayasu puede afectar a cualquier
segmento de la aorta y sus ramas vasculares y, por lo general, produce
inflamación de la raíz aórtica, lo que puede conducir a la dilatación del
aneurisma o al engrosamiento de la pared. El único tratamiento eficaz para la
regurgitación aórtica grave es la restauración de la competencia valvular, que
con frecuencia se logra reemplazando la válvula con una prótesis y la aorta
enferma con un injerto. Otras opciones, en centros con experiencia, incluyen el
reimplante de la válvula aórtica nativa dentro de un injerto aórtico (p. ej.,
la técnica de David-V) o el reemplazo de la válvula aórtica y los senos con la
válvula pulmonar del propio paciente junto con la colocación de un homoinjerto
pulmonar (es decir, el procedimiento de Ross). La retención de la válvula
aórtica nativa tiene ventajas sobre el reemplazo; las válvulas mecánicas son
duraderas pero requieren anticoagulación permanente, y las válvulas
bioprotésicas tienen una durabilidad limitada en adultos jóvenes y
probablemente requieran una nueva intervención dentro de los 10 años.
La toma de decisiones clínicas es compleja, porque la
naturaleza inflamatoria de la aortopatía de la paciente genera preocupación por
las complicaciones intraoperatorias relacionadas con la friabilidad del tejido,
así como por la posibilidad de una dehiscencia anastomótica posterior. La
consulta multidisciplinaria y el inicio de la terapia antiinflamatoria son
necesarios antes de la cirugía. La fracción de eyección del ventrículo
izquierdo reducida es preocupante, pero es poco probable que retrasar la
intervención por un período corto para permitir la supresión del proceso
inflamatorio aumente el riesgo de disfunción sistólica irreversible del
ventrículo izquierdo. Aunque no es eficaz para la regurgitación aórtica, la
terapia médica dirigida por las guías suele ser eficaz para la disfunción
sistólica del ventrículo izquierdo. Si el estado del paciente es demasiado
inestable para aplazar la intervención, la cirugía debe ser recomendada después
de una cuidadosa discusión riesgo-beneficio con la paciente y su familia.
EVOLUCIÓN
Debido a la preocupación por las complicaciones
perioperatorias, la intervención quirúrgica se retrasó 16 días, tiempo durante
el cual se inició terapia inmunosupresora, que incluía metilprednisolona
intravenosa en dosis de 500 mg diarios durante 5 días, seguida de prednisona
oral en dosis de 55 mg diarios (1 mg por kilogramo de peso corporal por día) y
la administración subcutánea semanal simultánea de 15 mg de metotrexato. No se
utilizaron inhibidores del factor de necrosis tumoral α dado el grado de
insuficiencia cardiaca. Se inició tratamiento médico para la insuficiencia
cardíaca, que incluía sacubitrilo-valsartán (24 mg de sacubitrilo y 26 mg de
valsartán dos veces al día) y espironolactona (12,5 mg al día). La paciente prefirió encarecidamente evitar la
anticoagulación a largo plazo, por lo que se realizó un reemplazo quirúrgico de
la raíz aórtica con conservación de la válvula (técnica de David-V) con
reparación concomitante de la válvula aórtica.
El examen patológico de la aorta resecada ( Figura 4 )
mostró una destrucción extensa de la túnica media con engrosamiento de la
íntima prominente y fibrosis adventicia pero sin inflamación activa. Después de
la operación, solo tuvo insuficiencia aórtica leve; con el tratamiento médico
dirigido por las guías para la insuficiencia cardíaca, la fracción de eyección
del ventrículo izquierdo aumentó y el volumen del ventrículo izquierdo anormalmente
alto disminuyó. Fue dada de alta el día 26 del hospital mientras recibía
sacubitrilo-valsartán (24 mg de sacubitrilo y 26 mg de valsartán dos veces al
día), espironolactona (25 mg al día) y empagliflozina (10 mg al día), con un
plan para aumentar las dosis como un paciente externo.

Figura 4. Fotomicrografías representativas de
Patología Quirúrgica.
Análisis histológico de la pieza quirúrgica después de
la tinción con coloración de Verhoeff–van Gieson, para resaltar las fibras
elásticas, que son un componente mayor de la túnica media (asterisco), mostró
segmentos distales normales de la aorta (Panel A), con un fino endotelio
(flecha), túnica media intacta, con adventicia sana, y con vasa vasorum (doble
cruz). Proximalmente había segmentos de destrucción casi total de la elástica
interna, la cual estaba asociada a prominente engrosamiento de la íntima (doble
flecha) (Panel B).
PONENTE
La ausencia de inflamación activa en el examen
patológico apoya la idea de que la terapia inmunosupresora preoperatoria para
la arteritis de Takayasu fue efectiva. La terapia antiinflamatoria puede
retirarse o interrumpirse si se produce la remisión, pero es esencial la
vigilancia a largo plazo con exámenes seriados del pulso, medición de
marcadores inflamatorios e imágenes transversales de la aorta y las ramas de
los vasos. La angiografía por resonancia magnética se puede utilizar con este
propósito para reducir la exposición a la radiación, y la FDG-PET se reserva
para los casos en los que existe preocupación acerca de la inflamación continua
o recurrente. Se necesita atención continua por parte de un equipo multidisciplinario,
que incluya un reumatólogo, un cardiólogo y un cirujano vascular.
COMENTARIO
La arteritis de Takayasu, que a menudo se manifiesta
inicialmente como síntomas inespecíficos y anomalías de laboratorio, presenta
un desafío diagnóstico. Después de que se desarrollaron artralgias, fatiga y febrícula,
se encontró que nuestra paciente tenía marcadores inflamatorios sustancialmente
elevados. A pesar de un estudio extenso, el diagnóstico se hizo solo después de
que ella presentó una insuficiencia cardíaca descompensada causada por
insuficiencia aórtica severa, lo que llevó a imágenes que mostraron dilatación
inflamatoria de la raíz aórtica.
La arteritis de Takayasu es una rara enfermedad
inflamatoria crónica de la aorta y sus ramas arteriales de causa aún
desconocida que ocurre con mayor frecuencia en mujeres que en hombres y que
suele manifestarse antes de los 40 años de edad. 1 Las estimaciones de
prevalencia varían ampliamente, de 0,9 casos por millón de personas en los
Estados Unidos 2 a 40 casos por millón en Japón. 3 Los primeros estudios de
autopsias en Japón indicaron una prevalencia de hasta 1 en 3000, lo que
respalda un subdiagnóstico sustancial. 4 La vasculitis por arteritis de
Takayasu se caracteriza por autoinmunidad mediada por células con infiltración
de linfocitos γδ, células dendríticas y granulocitos, aunque, como en este
caso, la inmunosupresión puede ocultar estos hallazgos, dejando solo evidencia
de destrucción medial.5 La fisiopatología probablemente implica una interacción
entre la genética y el medio ambiente. Las variantes en múltiples loci, en la
región HLA y en otros lugares, se han asociado con la arteritis de Takayasu,
pero no se ha identificado ninguna variante como causal. 6
Los síntomas constitucionales, que incluyen fatiga,
febrícula, sudores nocturnos y pérdida de peso, son manifestaciones tempranas
frecuentes de la arteritis de Takayasu y ocurren en hasta el 65% de los
pacientes en series de casos. 7,8 Aunque la sinovitis es rara, las artralgias
son comunes (en aproximadamente el 40% de los pacientes). 8 Las manifestaciones
dermatológicas como el eritema nodoso o una erupción malar fotosensible similar
al lupus pueden ser hallazgos tempranos, que ocurren en aproximadamente el 20%
de los pacientes. 9A medida que la enfermedad progresa, predominan los síntomas
de vasculitis de grandes vasos. Puede ocurrir sensibilidad de la arteria
carótida (carotidinia), soplos arteriales, hipertensión, presión arterial
discrepante, síntomas gastrointestinales, angina, trastornos visuales y síntomas
neurológicos, dependiendo de la distribución de la inflamación arterial y la
estenosis resultante. 1 En todas las etapas, pero especialmente en la fase
estenótica tardía de la enfermedad, los pulsos distales suelen estar
asimétricamente disminuidos o incluso ausentes, lo que lleva al apodo de
"enfermedad sin pulso". 10
Se han desarrollado varios criterios de diagnóstico,
en gran parte con fines de investigación. 11 Los criterios de clasificación del
American College of Rheumatology (ACR) de 1990 para la arteritis de Takayasu
incluyen la presencia de al menos tres de seis características: una edad de
inicio de menos de 40 años, claudicación, disminución del pulso de la arteria
braquial, una diferencia de presión arterial de más de 10 mm Hg entre brazos,
soplo subclavio o aórtico e imágenes aórticas anormales. Según los informes,
esta definición tiene una sensibilidad del 90,5 % y una especificidad del 97,8
% para la arteritis de Takayasu, aunque el criterio de la edad ha sido debatido
dado que entre el 13 y el 17,5 % de los pacientes reciben un diagnóstico
después de los 40 años. 11
La regurgitación aórtica complica aproximadamente el
25% de los casos de arteritis de Takayasu. La malacoaptación debida a la
dilatación de la raíz aórtica es probablemente el mecanismo principal. 1La
regurgitación aórtica grave se asocia clásicamente con un soplo diastólico
decreciente temprano, que se escucha mejor en el borde esternal inferior
izquierdo. Otros hallazgos incluyen un pulso saltón con aumento sistólico rápido
y colapso diastólico (pulso de golpe de ariete de Watson), balanceo de la
cabeza (signo de Musset) y enrojecimiento y palidez repetidos de los lechos
ungueales capilares (pulso de Quincke). Independientemente de la causa, si no
se trata, la insuficiencia aórtica grave produce dilatación progresiva del
ventrículo izquierdo, remodelación, disfunción sistólica, insuficiencia
cardíaca y, en última instancia, la muerte; es necesario reparar o reemplazar
la válvula para interrumpir esta historia natural. 12
En ausencia de ensayos aleatorizados, el manejo se
guía en gran medida por la opinión de expertos. El pilar del tratamiento es la
terapia con dosis altas de glucocorticoides, seguida de la adición de agentes
ahorradores de glucocorticoides. 13 Las guías del ACR siguen favoreciendo la
azatioprina, la leflunomida, el metotrexato, el micofenolato de mofetilo o la
ciclofosfamida, sobre la base de datos observacionales. 13 Sin embargo, el
tocilizumab, un anticuerpo monoclonal contra el receptor de la interleucina-6
que ha demostrado eficacia en la arteritis de células gigantes, también se ha
utilizado con eficacia. 14 En pacientes sometidos a cirugía aórtica, el
pretratamiento con inmunosupresión se utiliza de forma rutinaria con el
objetivo de reducir el riesgo de dehiscencia anastomótica, una complicación
catastrófica, 15aunque este enfoque no ha sido estudiado prospectivamente. El
momento adecuado de la intervención quirúrgica en relación con la terapia
inmunosupresora no está claro y las decisiones se individualizan de acuerdo con
la presentación clínica y la estabilidad del paciente.
Este caso destaca los desafíos de diagnóstico y manejo
presentados por la arteritis de Takayasu, una vasculitis de grandes vasos poco
estudiada y poco reconocida. Especialmente en mujeres jóvenes que presentan
síntomas constitucionales y marcadores inflamatorios elevados, es importante un
examen físico cuidadoso para detectar manifestaciones vasculares y valvulares
de este diagnóstico difícil de alcanzar.
Traducción de:
“The Young and the Breathless”
Jacob J. Mayfield, M.D., Danelle Hidano, M.D., J.
Alexander Torres, M.D., Mathilde H. Pioro, M.D., and Catherine M. Otto, M.D.
https://www.nejm.org/doi/full/10.1056/NEJMcps2202710?query=featured_home
Referencias
1. Mason JC. Takayasu arteritis — ad[1]vances
in diagnosis and management.
Nat Rev Rheumatol 2010;6:406-15.
2. Cotch M, Hoffman G. The prevalence,
epidemiology and cost of hospitalizations
for vasculitis in New York State — 1986
to 1990. Arthritis Rheum 1995;38:Suppl:
S225. abstract.
3. Toshihiko N. Current status of large
and small vessel vasculitis in Japan. Int J
Cardiol 1996;54:Suppl:S91-S98.
4. Nasu T. Takayasu’s truncoarteritis in
Japan: a statistical observation of 76 au[1]topsy
cases. Pathol Microbiol (Basel) 1975;
43:2-O:140-6.
5. Inder SJ, Bobryshev YV, Cherian SM,
et al. Immunophenotypic analysis of the
aortic wall in Takayasu’s arteritis: involve[1]ment
of lymphocytes, dendritic cells and
granulocytes in immuno-inflammatory
reactions. Cardiovasc Surg 2000;8:141-8.
6. Terao C. Revisited HLA and non-HLA
genetics of Takayasu arteritis — where
are we? J Hum Genet 2016;61:27-32.
7. Furuta S, Cousins C, Chaudhry A,
Jayne D. Clinical features and radiological
findings in large vessel vasculitis: are
Takayasu arteritis and giant cell arteritis
2 different diseases or a single entity?
J Rheumatol 2015;42:300-8.
8. Bicakcigil M, Aksu K, Kamali S, et al.
Takayasu’s arteritis in Turkey — clinical
and angiographic features of 248 patients.
Clin Exp Rheumatol 2009;27:Suppl 1:S59-
S64.
9. Sharma BK, Jain S, Sagar S. Systemic
manifestations of Takayasu arteritis: the
expanding spectrum. Int J Cardiol 1996;
54:Suppl:S149-S154.
10. Shimizu K, Sano K. Pulseless disease.
J Neuropathol Clin Neurol 1951;1:37-47.
11. de Souza AWS, de Carvalho JF. Diag[1]nostic
and classification criteria of Takaya[1]su
arteritis. J Autoimmun 2014;48-49:79-83.
12. Otto CM, Nishimura RA, Bonow RO,
et al. 2020 ACC/AHA guideline for the
management of patients with valvular
heart disease: a report of the American
College of Cardiology/American Heart
Association Joint Committee on Clinical
Practice Guidelines. Circulation 2021;
143(5):e72-e227.
13. Maz M, Chung SA, Abril A, et al. 2021
American College of Rheumatology/Vas[1]culitis
Foundation guideline for the man[1]agement
of giant cell arteritis and Takaya[1]su
arteritis. Arthritis Rheumatol 2021;73:
1349-65.
14. Singh A, Danda D, Hussain S, et al.
Efficacy and safety of tocilizumab in treat[1]ment
of Takayasu arteritis: a systematic
review of randomized controlled trials.
Mod Rheumatol 2021;31:197-204.
15. Matsuura K, Ogino H, Kobayashi J,
et al. Surgical treatment of aortic regurgi[1]tation
due to Takayasu arteritis: morbidity and mortality. Circulation 2005;112:3707-12.