Un hombre de 57 años fue evaluado en la consulta de
gastroenterología de este hospital por ascitis de aparición reciente.
Veinte años antes de la presentación actual, el
paciente comenzó a tener hinchazón de las piernas, que inicialmente se
presentaba solo después de los viajes en avión. Durante la quinta década de la
vida, la tumefacción de las piernas se presentaba a diario. Trece años antes de
la presentación actual, el paciente comenzó a tener evacuaciones intestinales
frecuentes (hasta cuatro por día) con heces sueltas malolientes y aumento de
flatos. Fue evaluado en la consulta de gastroenterología de este hospital. En
el examen, el abdomen estaba blando y sin dolor, sin evidencia de
organomegalia. Había edema con fóvea en ambas piernas. El nivel de proteína
total en sangre fue de 5,5 g por decilitro (rango de referencia, 6,0 a 8,3), y
el nivel de albúmina en sangre fue de 3,1 g por decilitro (rango de referencia,
3,3 a 5,0). Los resultados de otras pruebas de función hepática, incluidos los
niveles de enzimas hepáticas, fueron normales. El nivel de proteína en una
muestra de orina de 24 horas fue normal. El nivel en sangre de alfa 1
-antitripsina fue de 173 mg por decilitro (valor de referencia, <54). Los
niveles sanguíneos de IgG, IgA e IgM eran bajos. Las pruebas serológicas para
anticuerpos anti-transglutaminasa tisular y anticuerpos anti-giardia fueron
negativas.
La enterografía por tomografía computarizada (TC) del
abdomen y la pelvis, realizada después de la administración de material de
contraste intravenoso y oral, reveló engrosamiento mural difuso e
hipercaptación del intestino delgado, sin evidencia de estenosis u obstrucción (
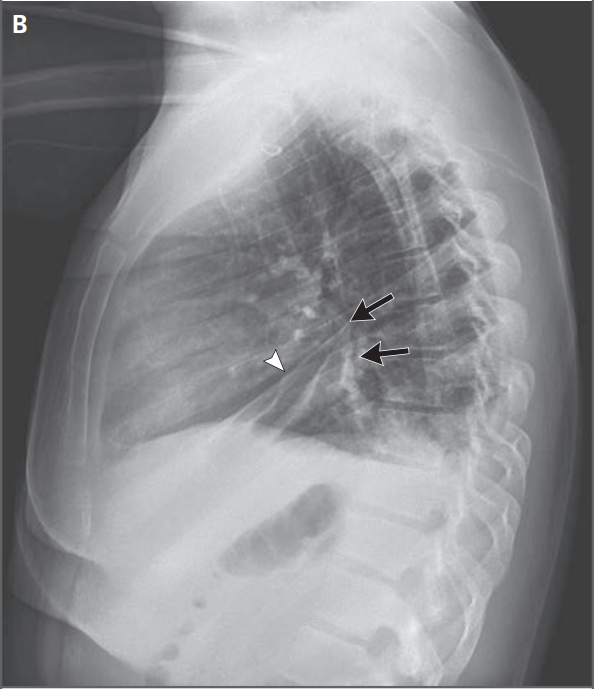
Figura 1A y 1B ). Además, había trazas de ascitis ( Figura 1B ).
Figura 1. Estudios de Imagen Inicial.
Trece años antes de la presentación actual, se realizó
una enterografía por TC del abdomen y la pelvis después de contraste
intravenoso y oral. Imágenes de reconstrucción axial y coronal (Paneles A y B, respectivamente)
muestran engrosamiento mural difuso e hiperrealce del intestino delgado
(Paneles A y B, flechas) y trazas de ascitis entre asas (Panel B, punta de
flecha). En la presentación actual, la TC de tórax, abdomen y pelvis después de contraste intravenoso y oral, las
imágenes axiales (Paneles C a F) muestran múltiples lesiones hepáticas focales
hipocaptantes nuevas (Paneles C y D, flechas), engrosamiento mural difuso del
intestino delgado. hiperrealce (Panel E, flecha) similar a los hallazgos
observados en el estudio anterior, ascitis de volumen moderado (Panel E, punta
de flecha) y linfadenopatía subcarinal (Panel F, flecha).
La esofagogastroduodenoscopia (EGD) y la colonoscopia
no revelaron anomalías visibles. Una muestra de biopsia aleatoria del duodeno
mostró un área focal con más de 70 eosinófilos por campo de gran aumento; una
muestra de biopsia del antro gástrico mostró eosinófilos dispersos en la lámina
propia. Se hizo un diagnóstico de trabajo de enteropatía con pérdida de
proteínas, aunque ni un curso de 2 semanas de prednisona oral ni un curso de 6
semanas de budesonida oral condujeron a una disminución en el número de
deposiciones o edema de la pierna. No se siguió ningún tratamiento adicional.
Once años antes de la presentación actual, el paciente
fue evaluado en la consulta de gastroenterología de este hospital. No había
habido pérdida de peso ni infecciones recurrentes, pero persistía el edema de
las piernas. La repetición de la EGD no reveló anomalías visibles, y una
muestra de biopsia aleatoria del duodeno mostró menos eosinófilos que la
muestra anterior, sin daño epitelial. La enterografía por TC reveló un
engrosamiento difuso de la pared de todo el intestino delgado y trazas de
ascitis. Se recomendó el tratamiento para la enteropatía con pérdida de
proteínas, que incluía limitar el consumo de sal, aumentar las proteínas en la
dieta y usar medias de compresión. Durante los siguientes 10 años, el edema de
la pierna se mantuvo estable y ocasionalmente se trató con agentes diuréticos,
lo que no condujo a una mejoría sustancial.
Seis meses antes de la presentación actual, el
paciente comenzó con disnea de esfuerzo. Tres meses antes de esta presentación,
desarrolló distensión abdominal. El paciente inicialmente atribuyó la disnea y
la distensión abdominal al aumento de peso debido a la reducción de la
actividad física durante la cuarentena por la pandemia de la enfermedad por
coronavirus 2019 (Covid-19). Seis semanas antes de esta presentación, el
paciente fue evaluado por su médico de atención primaria en una
videoconferencia. Sobre la base de esta evaluación, se obtuvieron estudios de
laboratorio y de imágenes. El nivel de proteína total en sangre fue de 4,5 g
por decilitro (rango de referencia actualizado, 6,4 a 8,3) y el nivel de
albúmina en sangre fue de 2,1 g por decilitro. El hemograma completo, los
niveles sanguíneos de electrolitos y glucosa, y los resultados de las pruebas
de función hepática y renal fueron normales.
La tomografía computarizada del tórax, realizada
después de la administración de contraste intravenoso, reveló múltiples nódulos
pulmonares pequeños, ganglios linfáticos mesentéricos superiores y subcarinales
agrandados y ascitis de volumen moderado en la parte superior del abdomen.
Tres semanas después, se realizó una paracentesis y se
extrajeron 3,1 litros de líquido ascítico de aspecto lechoso. Los resultados
del análisis del líquido ascítico se muestran en la Tabla 1 . El examen
citológico del líquido ascítico no reveló células malignas. El paciente fue
remitido a la clínica de gastroenterología de este hospital para una evaluación
adicional.
Tabla 1. Análisis de líquido ascítico.
En la presentación actual, el paciente informó que
tenía deposiciones con heces formadas normalmente y sin dolor abdominal. Su
esposa había notado que parecía moverse lentamente cuando realizaba actividades
de la vida diaria y que había perdido masa muscular en los brazos. No había
antecedentes de cirugía abdominal ni traumatismo en el abdomen. Otros
antecedentes médicos incluyen diabetes mellitus tipo 2, psoriasis,
osteoartritis e hipogammaglobulinemia. No había antecedentes de infecciones
recurrentes, asma o dermatitis atópica. Los medicamentos incluyeron aspirina,
insulina glargina y metformina. No se conocían alergias a medicamentos. El
paciente vivía con su esposa en un suburbio de Boston y trabajaba en una
oficina. No fumó durante toda su vida, bebía alcohol ocasionalmente y no
consumía drogas ilícitas. Su madre tenía cáncer de colon.
El examen físico fue limitado, porque la evaluación se
hizo en una videoconferencia en el contexto de la pandemia de Covid-19. Se
obtuvieron estudios de imagen.
La tomografía computarizada del abdomen y la pelvis,
realizada después de la administración de material de contraste intravenoso y
oral, reveló múltiples lesiones hipocaptantes nuevas en los lóbulos hepáticos
derecho e izquierdo, engrosamiento difuso de la pared del intestino delgado,
ascitis de volumen moderado, várices abdominales superiores , y agrandamiento
de los ganglios pélvicos y abdominales superiores ( Figura 1C, 1D y 1E). Las
únicas lesiones hepáticas que eran lo suficientemente grandes para un muestreo
percutáneo confiable se encontraban en la zona craneal lejana de la cúpula
hepática, donde el movimiento respiratorio y el cruce del diafragma habrían
hecho que un intento de biopsia guiada por TC o ultrasonográfica fuera muy
desafiante, si no prohibitivo. La TC de tórax reveló nódulos en árbol en brote
y agrupados, predominantemente en los lóbulos inferiores dependientes, y
adenopatías hiliares y subcarinales derechas ( Figura 1F ).
Las pruebas de anticuerpos del virus de la hepatitis C
y de anticuerpos de superficie del virus de la hepatitis B, antígeno de superficie
y anticuerpos core fueron negativas. Se repitió la paracentesis y se extrajeron
3,2 litros de líquido ascítico de aspecto lechoso. Los resultados del análisis
del líquido ascítico se muestran en la Tabla 1. Una tinción ácido-resistente de
una muestra concentrada de líquido ascítico fue negativa. El examen citológico
del líquido ascítico no reveló células malignas. También se realizó una biopsia
hepática transyugular guiada por fluoroscopia. El gradiente de presión venoso
hepático (GPVH) fue de 2 mmHg (valor normal, <5); la presión de la aurícula
derecha era de 7 mm Hg, la presión venosa hepática en cuña de 11 mm Hg y la
presión venosa hepática libre de 9 mm Hg.
El examen histológico de la muestra de biopsia
hepática reveló inflamación portal mixta leve y esteatosis de grado 1 sin esteatohepatitis.
Una tinción tricrómica mostró edema leve del tracto portal pero sin fibrosis.
Los cultivos de micobacterias del líquido ascítico
fueron negativos. Se realizó una prueba diagnóstica.
Diagnóstico diferencial
Este hombre de 57 años con un diagnóstico previo de
gastroenteritis eosinofílica presentó un inicio relativamente nuevo de ascitis
quilosa de rápida acumulación. Tenía signos de atrofia muscular y reportó una
dificultad creciente para realizar actividades de la vida diaria. Al
desarrollar un diagnóstico diferencial inicial, se destacan dos características
prominentes de la presentación de este paciente: el diagnóstico previo de
enteropatía con pérdida de proteínas en presencia de enfermedad inflamatoria
eosinofílica y la ascitis quilosa nueva y que empeora rápidamente. La primera
pregunta a abordar es si el desarrollo de ascitis quilosa está relacionado con
la enfermedad inflamatoria eosinofílica.
GASTROENTERITIS EOSINOFÍLICA
La gastroenteritis eosinofílica es una enfermedad
inflamatoria eosinofílica del intestino. Los síntomas comunes incluyen dolor
abdominal, náuseas, vómitos y diarrea. 1 Puede ocurrir enteropatía con pérdida
de proteínas, que estaba presente en este paciente. La ascitis es una rara
manifestación de la enfermedad. Este paciente tenía un curso latente de
gastroenteritis eosinofílica que había durado casi 15 años, lo cual es típico
de esta enfermedad de progresión lenta. Sin embargo, su presentación actual con
acumulación rápidamente progresiva de ascitis quilosa sugiere un nuevo proceso
que probablemente no esté relacionado con el diagnóstico previo de
gastroenteritis eosinofílica. Por tanto, centraré el diagnóstico diferencial en
las causas de la ascitis quilosa.
ASCITIS QUILOSA
La ascitis quilosa es una forma rara de ascitis que se
caracteriza por la acumulación de líquido linfático rico en proteínas y
quilomicrones en la cavidad peritoneal. La linfa se genera a partir del líquido
que se extrae de la vasculatura, se filtra a través de los tejidos y luego se
recolecta en los conductos linfáticos. En la mucosa gastrointestinal del
intestino delgado, este líquido contiene quilomicrones que son el principal mecanismo
para el transporte de ácidos grasos de cadena larga desde los alimentos
digeridos. La presencia de quilomicrones le da al líquido linfático intestinal
un aspecto blanco lechoso, que también se puede observar en la ascitis quilosa.
La ascitis quilosa es distinta de otros tipos más comunes de ascitis que a
menudo se clasifican con el uso del gradiente de albúmina sero-ascítico (SAAG).
La ascitis asociada con un SAAG alto (>1,1) se observa a menudo en pacientes
con hipertensión portal debido a cirrosis, pero también se puede observar en
pacientes con insuficiencia cardíaca. La ascitis asociada a un SAAG bajo es
típica de estados inflamatorios.
Puede ser útil dividir las causas de la ascitis
quilosa en dos categorías fisiológicas ( Figura 2 ). La primera categoría
incluye condiciones que causan la interrupción de la vasculatura linfática, lo
que resulta en una fuga de linfa desde el sistema linfático hacia el espacio
peritoneal. La interrupción linfática generalmente es causada por daño en los
conductos por cirugía abdominal; el trauma no quirúrgico es una causa menos
común. 2Este paciente no tenía antecedentes de cirugía abdominal o trauma
reciente o remoto en el abdomen. La segunda categoría incluye condiciones que
causan obstrucción de la vasculatura linfática. La obstrucción linfática puede
ocurrir a través del bloqueo directo de los vasos linfáticos o de un drenaje
deficiente en el sistema venoso, el último de los cuales puede ser causado por
insuficiencia cardíaca congestiva o hipertensión portal debida a cirrosis.
Figura 2. Causas de la ascitis quilosa.
HIPERTENSIÓN PORTAL
La hipertensión portal es una causa frecuente de
ascitis quilosa que no está relacionada con lesión linfática. En el momento de
nuestra evaluación inicial, aún no habíamos medido el HVPG (hepatic venous
pressure gradient), lo que finalmente descartó hipertensión portal en este
paciente. 3,4
En pacientes con cirrosis, la hipertensión portal
aumenta la tensión de cizallamiento (shear stress), en los capilares
esplácnicos, lo que aumenta la liberación de óxido nítrico que produce vasodilatación
sistémica. La vasodilatación sistémica activa los barorreceptores, que a su vez
activan el sistema renina-angiotensina-aldosterona, lo que provoca la retención
de sal y agua. El líquido puede acumularse en el espacio peritoneal, generando
ascitis baja en proteínas. Por razones que no están claras, en una pequeña
proporción de pacientes con cirrosis se desarrolla ascitis quilosa. Una posible
explicación es que el aumento de la presión venosa impide un drenaje linfático
eficaz hacia las venas, lo que provoca hipertensión linfática y pérdida de
linfa hacia la cavidad peritoneal.
Sobre la base de la información que teníamos antes de
realizar la biopsia hepática, ¿podría ser la hipertensión portal debida a
cirrosis una posible causa de ascitis quilosa en este paciente? Algunas de las
características de las imágenes, incluidas las várices abdominales superiores,
sugieren hipertensión portal, pero estos hallazgos no son específicos,
particularmente porque el paciente no tenía factores de riesgo claros para la
cirrosis. Tenía pruebas negativas para virus de hepatitis; no se realizaron
pruebas serológicas para enfermedades autoinmunes. Tenía diabetes tipo 2, pero
no tenía ninguna otra característica del síndrome metabólico que lo pusiera en
mayor riesgo de esteatohepatitis. Bebía alcohol con poca frecuencia, por lo que
es poco probable que padezca una enfermedad hepática debido al consumo de
alcohol. Los resultados de sus pruebas de laboratorio fueron notables por
hipoalbuminemia, que puede ocurrir con la cirrosis, pero es más probable que se
explique por su diagnóstico conocido de enteropatía con pérdida de proteínas.
Aunque sus estudios de imágenes hepáticas mostraron anormalidades, las lesiones
identificadas no eran típicas de cirrosis y eran más sugestivas de un proceso
infiltrativo.
OBSTRUCCIÓN LINFÁTICA
Incluso antes de que se realizara la biopsia hepática
y se midió el HVPG (lo que descartó definitivamente la cirrosis), el paciente
no tenía evidencia para respaldar un diagnóstico de cirrosis u otras causas de
congestión venosa, y el diagnóstico diferencial se redujo a otras causas obstructivas
de congestión linfática. Estos se pueden agrupar como causas inflamatorias, infecciosas
y relacionadas con cáncer.
Las causas inflamatorias de obstrucción linfática
incluyen sarcoidosis, exposición a radiación y fibrosis peritoneal. El paciente
no tenía otros signos de estas condiciones inflamatorias, por lo que son
diagnósticos poco probables en este caso. Asimismo, no presentaba factores de riesgo de causas
infecciosas de obstrucción linfática, como filariasis o enfermedad
micobacteriana, aunque la ausencia de factores de riesgo no permite descartar
estas infecciones. Por lo tanto, el cáncer parece ser la causa más probable de
una enfermedad infiltrativa del hígado que ha provocado obstrucción linfática y
ascitis quilosa en este paciente. Aunque no tenía un cáncer primario conocido,
los linfomas de células B a menudo se manifiestan con enfermedad linfática, son
una causa importante de ascitis quilosa y también podrían explicar las
anomalías hepáticas observadas en la TC. 4Los linfomas de células B pueden ser
agresivos, con un crecimiento rápido y un empeoramiento de los síntomas durante
un período de algunas semanas, lo que sería compatible con la enfermedad de
este paciente.
Varias estrategias de diagnóstico pueden ayudar a
establecer el diagnóstico de linfoma de células B. Estos incluyen la toma de
muestras de líquido ascítico, así como la biopsia del mesenterio o de los
ganglios linfáticos mesentéricos, la mucosa gastrointestinal o el hígado. En
este paciente, no se identificaron células malignas en el líquido ascítico y la
tinción ácido-resistente y los cultivos para micobacterias fueron negativos.
Finalmente optamos por realizar una biopsia de hígado. Elegimos un abordaje
transyugular porque las lesiones hepáticas detectadas en la TC eran demasiado
pequeñas o estaban demasiado cerca del diafragma para poder tomar muestras de
manera confiable y segura con una biopsia guiada; además, un abordaje
transyugular permitiría medir el HVPG. Decidimos no realizar más pruebas de
imagen, como la tomografía por emisión de positrones con 18
F-fluorodesoxiglucosa (FDG-PET), sin un diagnóstico histológico.
Desafortunadamente, la biopsia hepática transyugular
no dio como resultado un diagnóstico definitivo, probablemente porque no
pudimos obtener muestras de tejido adecuadas. 5 Las muestras de biopsia
generalmente eran pequeñas y no estaban dirigidas a las anomalías observadas en
la TC, porque la biopsia no se realizó con la guía de la TC. El linfoma de
células B permaneció en la parte superior del diagnóstico diferencial, por lo
que el paciente se sometió a continuación a una biopsia hepática transabdominal
guiada por TC, lo que permitió que el especialista en procedimientos intentara
visualizar el área de interés objetivamente.
DIAGNÓSTICO CLÍNICO PRESUNTIVO DE LA SALA
LINFOMA DE CÉLULAS B.
PRUEBAS DE DIAGNÓSTICO
El paciente se sometió a una biopsia hepática focal
percutánea guiada por TC aproximadamente 1 mes después de la presentación. En
comparación con las imágenes de TC obtenidas en la presentación, las imágenes
axiales de TC obtenidas para el procedimiento (sin la administración de
material de contraste intravenoso) mostraron numerosas lesiones hepáticas
nuevas o agrandadas, mal definidas e hipocaptantes ( Figura 3A y 3B ). Una de
estas lesiones, en el segmento hepático 3, fue objeto de una biopsia central (
Figura 3B ).
Figura 3. Estudios de imagen posteriores.
Aproximadamente 1 mes después de la presentación, TC
sin contraste intravenoso fue para guiar la biopsia hepática focal percutánea.
Las imágenes axiales de TC (Paneles A y B) muestran numerosas lesiones
hepáticas nuevas, aumentadas de tamaño, mal definidas e hipocaptantes (Panel A,
flecha). Aproximadamente 1 mes después de la presentación, TC tras la administración
de contraste intravenoso y una tomografía por emisión de positrones FDG-PET con 18F-fluorodesoxiglucosa de abdomen y
pelvis. Una imagen de TC axial (Panel C)
muestra numerosas lesiones hepáticas hipocaptantes mal definidas (flecha). Una
proyección coronal de intensidad máxima (MIP) FDG-PET reconstrucción (Panel D)
muestra que las lesiones tienen intensa captación de FDG (flecha).
Aproximadamente 2 meses después de la presentación y 1 mes después del inicio
del tratamiento, se repitió la TC y FDG-PET a través del abdomen y pelvis. Una
imagen de TC axial (Panel E) muestra una disminución en el tamaño y número de lesiones
hepáticas focales (flecha). Un FDG-PET coronal MIP reconstrucción (Panel F)
muestra la resolución de la anormalidad correspondiente (flecha)
DISCUSIÓN PATOLÓGICA
El examen microscópico de la muestra obtenida en una
biopsia de hígado dirigida guiada por TC reveló borramiento por un infiltrado
difuso de células linfoides pleomórficas de tamaño mediano con cromatina
dispersa y nucléolos variablemente prominentes en un fondo de numerosos
macrófagos de cuerpo tingible y abundantes desechos apoptóticos ( Figura 4 ).
La tinción inmunohistoquímica de la muestra de biopsia reveló láminas de
células B CD20+ con coexpresión de CD10 y BCL6 y sin expresión de MUM1,
hallazgos consistentes con un subtipo similar a las células B del centro
germinal, de acuerdo con el algoritmo de Hans. 6Una inmunotinción específica
para MYC mostró una tinción irregular en aproximadamente el 40 % de los núcleos,
y una inmunotinción para BCL2 fue negativa. Una tinción inmunohistoquímica con
Ki-67 mostró un índice de proliferación elevado de aproximadamente 70%. La
hibridación in situ para el ARN codificado por el virus de Epstein-Barr fue
negativa.
Las características histológicas fueron intermedias
entre el linfoma difuso de células B grandes (DLBCL) y el linfoma de Burkitt y
fueron más consistentes con una caracterización de alto grado. La hibridación
fluorescente in situ (FISH) realizada en tejido fijado en formalina e incluido
en parafina no mostró un reordenamiento de MYC , BCL2 o BCL6 ; estos hallazgos
descartan la posibilidad de linfoma de células B de alto grado (HGBL) con un
reordenamiento de MYC y un reordenamiento de BCL2 o BCL6 (o ambos), lo que se conoce
como linfoma de doble impacto.
Según la clasificación de la Organización Mundial de
la Salud de 2017, HGBL se define por la presencia de un reordenamiento de MYC ,
BCL2 o BCL6 (o una combinación de los mismos) o la presencia de características
morfológicas blastoides o características intermedias entre DLBCL y linfoma de
Burkitt en ausencia de de criterios para otros tipos de linfoma. 7 Nuestro
diagnóstico en este caso fue HGBL (high[1]grade
B-cell lymphoma), no especificado (NOS), que representa un grupo heterogéneo de
linfomas de células B maduras agresivos. Aunque el diagnóstico de HGBL, NOS, se
asocia a un alto grado de variabilidad interobservador, 8 Se pensó que este
paciente tenía características intermedias entre DLBCL (diffuse large B-cell
lymphoma), y el linfoma de Burkitt: la presencia de núcleos pleomórficos
generalmente se asocia con DLBCL, y células de tamaño mediano y signos de alto
recambio celular, incluidos numerosos macrófagos de cuerpo tingible, abundantes
desechos apoptóticos y un elevado índice de proliferación Ki-67, se asocian
típicamente con el linfoma de Burkitt. Además, aunque el inmunofenotipo de
HGBL, NOS, es variable, se ha encontrado que la mayoría de los casos son del
subtipo similar a las células B del centro germinal con expresión de CD10 y
BCL6, y estas características se observaron en este caso. 9-11 Con la integración
de los análisis morfológico, inmunofenotípico y FISH, se estableció un
diagnóstico final de HGBL, NOS.

Figura 4. Muestra de biopsia hepática.
Tinción con hematoxilina y eosina de una sección del espécimen
obtenido en biopsia hepática dirigida (Paneles A, B, y C) muestra borramiento
completo por láminas de células linfoides (Panel A) en un fondo de numerosos macrófagos
de cuerpo visible (Panel B, puntas de flecha) y abundantes restos apoptóticos
(Panel B, flechas). Las células foides son de tamaño mediano y pleomórficas,
tienen nucléolos variablemente prominentes y cromatina dispersa, y se mezclan
con los macrófagos de cuerpo tingible (Panel C, flecha) y desechos apoptóticos.La
tinción inmunohistoquímica (paneles D y
E) muestra numerosas células B CD20+ (Panel D) con coexpresión de CD10 (Panel
E), hallazgos consistentes con un subtipo de células B del centro germinal. Tinción
inmunohistoquímica Ki-67 (Panel F) muestra un índice de proliferación elevado
del 70%.
DIAGNÓSTICO PATOLÓGICO
LINFOMA DE CÉLULAS B DE ALTO GRADO-NOS (not
otherwise specified).
DISCUSIÓN DEL MANEJO
La clasificación en evolución de HGBL, NOS, hace que
la selección del tratamiento sea un desafío. Hay pocos estudios retrospectivos
y datos prospectivos limitados que aborden específicamente el tratamiento de
esta condición. El régimen R-CHOP (rituximab, ciclofosfamida, doxorrubicina,
vincristina y prednisona) sigue siendo el tratamiento estándar para DLBCL
durante más de 20 años. No está claro si R-CHOP es una terapia suficiente para
HGBL, NOS; algunos análisis retrospectivos sugieren que lo es y otros sugieren
lo contrario. 12,13El régimen DA-EPOCH-R (etopósido, prednisona, vincristina,
ciclofosfamida, doxorrubicina y rituximab con dosis ajustada) es una terapia
más intensiva que R-CHOP, con etopósido agregado y doxorrubicina, vincristina y
etopósido administrados en un 96- infusión continua de una hora para erradicar
rápidamente los tumores proliferativos. Este régimen ha sido efectivo en el
tratamiento del linfoma de Burkitt, para el cual R-CHOP es decididamente
insuficiente, 14 así como en el tratamiento de variantes de alto grado de
DLBCL, incluido el linfoma de doble golpe. 15 DA-EPOCH-R también se comparó
directamente con R-CHOP en el tratamiento de DLBCL, NOS, y los dos regímenes se
asociaron con resultados similares. dieciséis
Extrapolando estos datos, seleccionamos DA-EPOCH-R
para el tratamiento de HGBL, NOS, en este paciente. Tuvo una respuesta completa
después de dos ciclos de terapia y continuó teniendo una respuesta completa
después de seis ciclos en total. Ya han pasado 7 meses desde que completó la
quimioterapia. Continúa en seguimiento en la clínica de oncología, y nuestra
esperanza es que el linfoma subyacente se haya curado.
ESTUDIOS DE IMAGEN ADICIONALES
Aproximadamente 1 mes después de la presentación, la
TC y FDG-PET a través del abdomen y la pelvis revelaron numerosas lesiones
hepáticas mal definidas con hipocaptación intensa de FDG ( Figura 3C y 3D ).
Aproximadamente 2 meses después de la presentación y 1 mes después del inicio
del tratamiento, la TC y FDG-PET repetidas a través del abdomen y la pelvis
revelaron una disminución en el tamaño y el número de lesiones hepáticas
focales y la resolución de la captación anormal de FDG correspondiente ( Figura
3E y 3F ).
SEGUIMIENTO Y MANEJO DE LA ASCITIS QUILOSA
El primer paso en el manejo de la ascitis quilosa es
abordar la causa subyacente. Aunque el linfoma de este paciente ha estado en
remisión desde el tratamiento, la ascitis quilosa se ha estabilizado pero
persiste. Esto plantea la cuestión de si la ascitis quilosa fue causada por un
linfoma o por otro proceso. La asociación temporal de la ascitis quilosa con el
linfoma sugiere una conexión causal. Además, la condición clínica del paciente
estaba empeorando antes del tratamiento del linfoma y ahora parece haberse
estabilizado, lo que sugiere que el linfoma desempeñó un papel en el desarrollo
de la ascitis quilosa y puede haber causado daño permanente a sus vasos
linfáticos. Se consideraron explicaciones alternativas para la ascitis quilosa.
El hecho de que la obstrucción linfática pueda causar tanto ascitis quilosa
como enteropatía perdedora de proteínas 3,17sugirió la posibilidad de una
enfermedad primaria de los linfáticos (p. ej., linfangiectasia intestinal).
Aunque la obstrucción linfática ahora puede explicar parcialmente su enteropatía
con pérdida de proteínas, es poco probable que un solo proceso de enfermedad
condujera a la obstrucción linfática que causó la enteropatía con pérdida de
proteínas 15 años antes de causar ascitis quilosa.
Desde que el paciente completó el tratamiento para el
linfoma, su ascitis quilosa se ha manejado con paracentesis seriadas. Sin
embargo, la confianza en la paracentesis seriada pone al paciente en riesgo de
complicaciones del procedimiento y peritonitis infecciosa secundaria, que puede
poner en peligro la vida. La ascitis quilosa no cede con la administración de
diuréticos en la mayoría de los casos. Una prueba de diuréticos en este
paciente redujo la acumulación de líquido ascítico y la administración de
diuréticos de mantenimiento parece ser beneficiosa, porque el líquido ascítico
se acumula sustancialmente más rápido cuando se omiten las dosis de diuréticos.
La ascitis quilosa también se puede controlar reduciendo la producción de
quilomicrones, lo que reduce la producción de linfa. Las estrategias incluyen
la reducción de los ácidos grasos de cadena larga en la dieta, así como la
administración de un tratamiento farmacológico con orlistat, que evita que las
lipasas del intestino procesen los triglicéridos en ácidos grasos libres.3
Estas opciones se han discutido con el paciente y hay planes para probar estos
enfoques para reducir o eliminar la necesidad de paracentesis.
DIAGNÓSTICO ANATÓMICO
LINFOMA DE CÉLULAS B DE ALTO GRADO, NO ESPECIFICADO DE
OTRO MODO.
Fuente:
Case 14-2022: A 57-Year-Old Man with Chylous Ascites
Michael Dougan, M.D., Ph.D., Mark A. Anderson, M.D.,
Jeremy S. Abramson, M.D., and Megan J. Fitzpatrick, M.D.
https://www.nejm.org/doi/full/10.1056/NEJMcpc2115856
Referencias
1. Talley NJ, Shorter RG, Phillips SF,
Zinsmeister AR. Eosinophilic gastroen[1]teritis:
a clinicopathological study of pa[1]tients
with disease of the mucosa, muscle
layer, and subserosal tissues. Gut 1990;
31:54-8.
2. Weniger M, D’Haese JG, Angele MK,
Kleespies A, Werner J, Hartwig W. Treat[1]ment
options for chylous ascites after ma[1]jor
abdominal surgery: a systematic re[1]view.
Am J Surg 2016;211:206-13.
3. Bhardwaj R, Vaziri H, Gautam A,
Ballesteros E, Karimeddini D, Wu GY.
Chylous ascites: a review of pathogenesis,
diagnosis and treatment. J Clin Transl
Hepatol 2018;6:105-13.
4. Steinemann DC, Dindo D, Clavien PA,
Nocito A. Atraumatic chylous ascites: sys[1]tematic
review on symptoms and causes.
J Am Coll Surg 2011;212(5):899-905.e1.
5. Deeken-Draisey A, Rao SM, Yang G-Y.
Pathology in patients with chronic liver
disease: a practical approach to liver bi[1]opsy
interpretation in patients with acute
and chronic liver diseases. Clin Liver Dis
2020;24:361-72.
6. Hans CP, Weisenburger DD, Greiner
TC, et al. Confirmation of the molecular
classification of diffuse large B-cell lym[1]phoma
by immunohistochemistry using
a tissue microarray. Blood 2004;103:275-
82.
7. Swerdlow SH, Campo E, Harris NL,
et al., eds. WHO classification of tu[1]mours
of haematopoietic and lymphoid
tissues. Rev. 4th ed. Geneva: World
Health Organization, 2017 (https://
publications.iarc.fr/Book-And-Report
-Series/Who-Classification-Of-Tumours/
WHO-Classification-Of-Tumours-Of
-Haematopoietic-And-Lymphoid-Tissues
-2017).
8. Collinge BJ, Hilton LK, Wong J, et al.
Characterization of the genetic landscape
of high-grade B-cell lymphoma, NOS —
an LLMPP project. Hematol Oncol 2021;
39:Suppl 2:157-9. abstract.
9. Ok CY, Medeiros LJ. High-grade B-cell
lymphoma: a term re-purposed in the re[1]vised
WHO classification. Pathology 2020;
52:68-77.
10. Olszewski A, Kurt H, Evens AM. De[1]fining
and treating high-grade B-cell lym[1]phoma,
NOS. Blood 2021 September 15
(Epub ahead of print).
11. Hüttl KS, Staiger AM, Richter J, et al.
The “Burkitt-like” immunophenotype and
genotype is rarely encountered in diffuse
large B cell lymphoma and high-grade B
cell lymphoma, NOS. Virchows Arch 2021;
479:575-83.
12. Lin P, Dickason TJ, Fayad LE, et al.
Prognostic value of MYC rearrangement
in cases of B-cell lymphoma, unclassifi[1]able,
with features intermediate between
diffuse large B-cell lymphoma and
Burkitt lymphoma. Cancer 2012;118:1566-
73.
13. Perry AM, Crockett D, Dave BJ, et al.
B-cell lymphoma, unclassifiable, with fea[1]tures
intermediate between diffuse large
B-cell lymphoma and burkitt lymphoma:
study of 39 cases. Br J Haematol 2013;162:
40-9.
14. Roschewski M, Dunleavy K, Abramson
JS, et al. Multicenter study of risk-adapted
therapy with dose-adjusted EPOCH-R in
adults with untreated Burkitt lymphoma.
J Clin Oncol 2020;38:2519-29.
15. Dunleavy K, Fanale MA, Abramson JS,
et al. Dose-adjusted EPOCH-R (etoposide,
prednisone, vincristine, cyclophosphamide,
doxorubicin, and rituximab) in untreated
aggressive diffuse large B-cell lymphoma
with MYC rearrangement: a prospective,
multicentre, single-arm phase 2 study.
Lancet Haematol 2018;5(12):e609-e617.
16. Bartlett NL, Wilson WH, Jung S-H,
et al. Dose-adjusted EPOCH-R compared
with R-CHOP as frontline therapy for dif[1]fuse
large B-cell lymphoma: clinical out[1]comes
of the phase III Intergroup Trial
Alliance/CALGB 50303. J Clin Oncol 2019;
37:1790-9.
17. Umar SB, DiBaise JK. Protein-losing
enteropathy: case illustrations and clini[1]cal
review. Am J Gastroenterol 2010;105:
43-9.