 |
| Hospital Municipal "Dr. Ángel Pintos" de Azul |
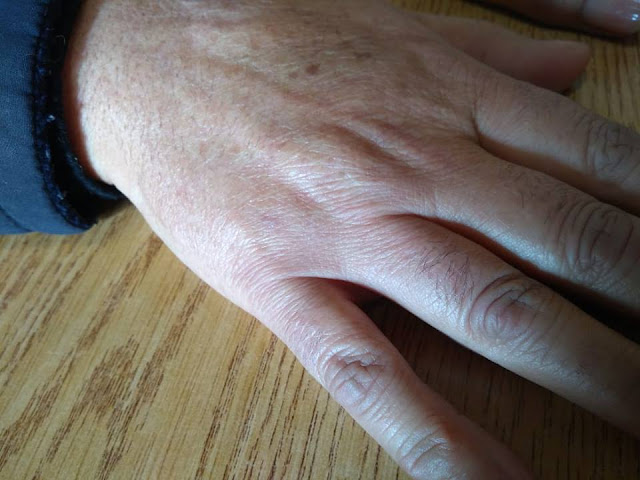 |
| Mano derecha sana |
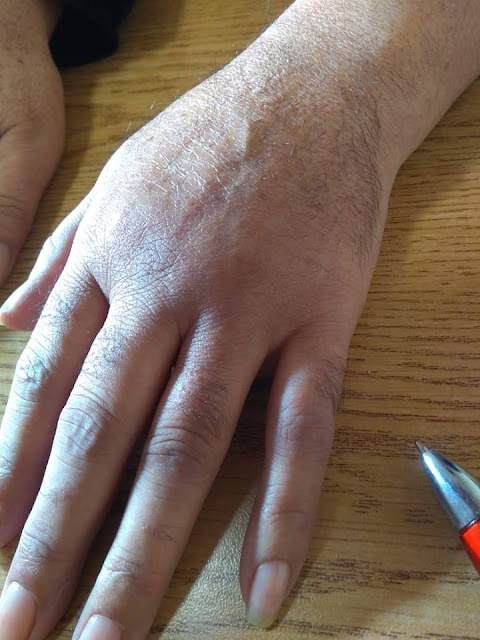 |
| Mano izquierda |
Mujer de 61 años con antecedentes de fractura de muñeca. Desarrolló dolor e impotencia funcional y rigidez de la mano cinco meses de evolución. Tiene edema duro. Cambios de coloración de la piel, y crecimiento del vello. Actualmente está en una etapa fría. Notar el crecimiento de pelo en mano izquierda
Presentó
Dr. Gustavo Rabazzano
Médico Especialista en Reumatología
Hospital Municipal de Azul
SÍNDROME DOLOROSO REGIONALCOMPLEJO
INTRODUCCIÓN
El síndrome de dolor regional
complejo (SDRC) es un trastorno de una región del cuerpo, generalmente de las
extremidades distales, que se caracteriza por dolor, hinchazón, rango de
movimiento limitado, inestabilidad vasomotora, cambios en la piel y
desmineralización ósea irregular. Con frecuencia comienza después de una
fractura, una lesión en los tejidos blandos o una cirugía.
DEFINICIÓN Y TERMINOLOGÍA
La definición de
consenso del SDRC es la siguiente [ 1 ]: "El SDRC describe una serie de
afecciones dolorosas que se caracterizan por un dolor regional continuo
(espontáneo o evocado) que es aparentemente desproporcionado en tiempo o grado
al habitual curso de cualquier trauma u otra lesión conocida. El dolor es
regional (no en un territorio específico del nervio o dermatoma) y generalmente
tiene un predominio distal de hallazgos anormales sensoriales, motores,
sudomotores, vasomotores y / o tróficos. El síndrome muestra progresión
variable a través del tiempo."
Existen muchos nombres alternativos más antiguos
para el SDRC en la literatura, incluida la distrofia simpática refleja, la
algodistrofia, la causalgia, la atrofia de Sudeck, la osteoporosis transitoria
y la atrofia aguda del hueso. La afección de las extremidades superiores
después de un accidente cerebrovascular o un infarto de miocardio a veces se
denominó "síndrome hombro-mano". En la era moderna, estos trastornos
se agrupan bajo el título único de CRPS [ 2 ].
Se han reconocido dos subtipos [ 1,2 ]:
● El tipo I (la forma también conocida como
distrofia simpática refleja) corresponde a pacientes con SDRC sin evidencia de
lesión del nervio periférico y representa aproximadamente el 90 por ciento de
las presentaciones clínicas.
● Tipo II anteriormente se denominaba
"causalgia" y se refiere a casos en los que la lesión del nervio
periférico está presente.
Sin embargo, la base fisiopatológica y la utilidad
clínica de la distinción entre estos dos subtipos de SDRC es incierta [ 1,3-5
].
Algunos investigadores también reconocen subtipos
"cálidos" y "fríos" de SDRC [ 6 ]:
● El SDRC "cálido" se distingue por el
aumento de la temperatura de la piel al inicio de los síntomas, sugiriendo
además que es un tipo inflamatorio
● El SDRC "frío" se diferencia por la
disminución de la temperatura de la piel al inicio de los síntomas
El término "dolor mantenido de forma
simpática" se considera un fenómeno variable asociado con una variedad de
trastornos, incluidos los tipos I y II de SDRC.
PATOGENIA : la patogenia del SDRC es desconocida.
Los mecanismos propuestos incluyen inflamación clásica, inflamación neurogénica
y cambios desadaptativos en la percepción del dolor a nivel del sistema
nervioso central [ 7 ].
Varios estudios sugieren que los pacientes con SDRC
tienen aumentos significativos en las citoquinas proinflamatorias (p. Ej.,
IL-1beta, IL-2, IL-6 y TNF-alfa) en el tejido afectado, así como en el plasma y
el líquido cefalorraquídeo [ 8-11 ] Entre los mecanismos propuestos para el
dolor persistente y la alodinia que son un sello distintivo del SDRC se
encuentran la liberación de mediadores inflamatorios y péptidos productores de
dolor por los nervios periféricos [ 7,12,13].] Los neuropéptidos que producen
dolor y signos de inflamación cuando se administran experimentalmente incluyen
sustancia P, neuropéptido Y y péptido relacionado con el gen de calcitonina. En
un proceso conocido como inflamación neurogénica, los impulsos nerviosos que se
propagan de forma antidrómica (es decir, de proximal a distal en un axón
nervioso sensitivo, particularmente nociceptivo) podrían conducir a la
liberación de neuropéptidos. También puede haber diafonía entre nervios
aferentes y eferentes en un sitio de lesión nerviosa. Este modelo también ofrece
una posible explicación para el fenómeno de la alodinia, en el que un estímulo
normalmente indoloro a la extremidad afectada, como el tacto ligero, produce
dolor significativo.
Otra posible explicación del dolor y la alodinia en
el SDRC es la sensibilización central, que aumenta la actividad en aferentes
nociceptivas debido a estímulos nocivos periféricos, daño tisular o lesión
nerviosa que conduce a una mayor transmisión sináptica en las neuronas
somatosensoriales del asta dorsal de la médula espinal [ 7 ] .
El papel del sistema nervioso simpático en SDRC no
está claro; sin embargo, las manifestaciones autonómicas previamente atribuidas
a la hiperactividad simpática podrían deberse a la hipersensibilidad a la
catecolamina [ 14 ] y pueden implicar la formación de un arco reflejo después
de un evento incitador [ 4 ]. El arco sigue las rutas del sistema nervioso
simpático y está modulado por centros corticales para producir alteraciones
vasculares periféricas. La sensación de dolor en respuesta a una lesión puede
conducir a una mayor sensibilidad de los axones lesionados a la epinefrina y a
otras sustancias liberadas por los nervios simpáticos locales. La sensibilidad
mejorada puede bloquearse mediante la administración intravenosa de agentes
simpaticolíticos.
El posible papel del sistema nervioso central en la
etiología del SDRC se basa en estudios que sugieren una reorganización cortical
en regiones sensoriales y motoras del cerebro [ 15-18 ], que podrían ser el
resultado de la actividad persistente de las neuronas nociceptivas primarias [
7 ].
Aunque los datos son limitados, los factores
genéticos también pueden estar involucrados en la patogénesis del SDRC. Un
estudio de casos y controles informó una frecuencia significativamente mayor de
HLA-DQ1 entre los pacientes afectados por SDRC tipo I [ 19 ]. Los resultados de
otro estudio sugirieron que el fenotipo de SDRC que progresó para desarrollar
distonía multifocal o generalizada se asoció con HLA-DR3 [ 20 ].
EPIDEMIOLOGÍA
El SDRC es más común en mujeres, con
una proporción de mujeres a hombres de 2: 1 a 4: 1 [ 21-24 ]. La incidencia
parece ser más alta en mujeres posmenopáusicas [ 23 ].
Eventos que incitan - Los eventos que incitan más
comunes que conducen a SDRC son fracturas, lesiones por aplastamiento,
esguinces y cirugía. Sin embargo, no se identificaron factores desencadenantes
en 0 a 10 por ciento de los pacientes [ 22,23 ]. En un estudio retrospectivo
basado en la población de 74 casos de SDRC, los eventos incitantes más
frecuentes fueron fractura y esguince, que se observaron en 46 y 12 por ciento
de los casos, respectivamente [ 22 ]. Varios otros factores precipitantes, como
aplastamiento, accidente cerebrovascular y contusión, estuvieron presentes en
el 42 por ciento restante de los casos.
Varios informes anteriores sugirieron que los
problemas psicosociales y los rasgos de personalidad actuaban como factores
predisponentes para CRPS [ 25-28 ]. Sin embargo, esta noción sigue siendo
controvertida porque los estudios posteriores no han confirmado dicha
asociación [ 29-31 ].
MANIFESTACIONES CLÍNICAS
Los principales síntomas
clínicos del SDRC son dolor, cambios sensoriales, alteraciones motoras,
síntomas autonómicos y cambios tróficos en la extremidad afectada [ 32 ]. El
inicio de SDRC generalmente ocurre dentro de cuatro a seis semanas del evento
incitador [ 32,33 ]. Los síntomas iniciales generalmente incluyen dolor,
eritema e hinchazón de la extremidad afectada [ 12 ]. En la mayoría de los
casos, la extremidad es cálida inicialmente, aunque algunos son fríos en la
presentación o evolucionan de cálidos a fríos. El SDRC en adultos ocurre más
comúnmente en los miembros superiores [ 22 ]. La participación de ambos
miembros superiores e inferiores en el mismo paciente es inusual [ 34]] Sin
embargo, los síntomas del SDRC pueden extenderse a lo largo del tiempo e
involucrar áreas adyacentes de la extremidad afectada o, ocasionalmente, otras
extremidades ipsilaterales o contralaterales [ 35,36 ].
Dolor : el dolor es, por lo general, el síntoma más
prominente y debilitante del SDRC. El dolor del SDRC se describe como una
sensación de ardor, escozor o desgarro que se siente en el interior de la
extremidad en la mayoría de los casos, aunque puede ser superficial en algunos
[ 27 ]. El dolor es más continuo, pero puede ser paroxístico. El dolor puede
verse exacerbado por el movimiento de las extremidades, el contacto, la
variación de temperatura o el estrés [ 32 ].
Sensorial : varios tipos de anormalidades
sensoriales son comunes en SDRC. Algunos pacientes tienen evidencia de
hiperalgesia, alodinia o hipoestesia en el examen [ 27 ]. Las alteraciones
sensoriales suelen ser distales en la extremidad, a veces en un patrón de media
/ guante .
Motor : casi todos los pacientes con SDRC tienen
alteraciones motoras funcionales relacionadas con el dolor [ 27,32 ]. El
movimiento de las extremidades puede estar limitado por edema, dolor o
contracturas. Algunos pacientes desarrollan manifestaciones motoras centrales
como temblores, mioclonos o posturas distónicas.
Autonómico : las diferencias en la temperatura de la
piel, el color de la piel, el sudor o el edema (que compara el lado afectado
con el lado no afectado) son síntomas comunes del SDRC [ 32 ]. Es de destacar
que el edema puede ser el resultado de una inflamación y / o disfunción
autonómica.
Trófico : los cambios tróficos que afectan el tejido
conectivo en el SDRC pueden incluir un aumento en el crecimiento del pelo,
aumento o disminución del crecimiento de las uñas, contracción y fibrosis de
las articulaciones y la fascia, y atrofia de la piel [ 32 ].
Etapas : aunque algunos informes iniciales
describieron tres etapas clínicas secuenciales de SDRC [ 37-42 ], este concepto
ha sido abandonado en gran parte por la mayoría de los expertos por la falta de
pruebas que respalden la existencia de etapas discretas [ 3,43 ]. Sin embargo,
la estadificación aún se considera útil por una proporción cada vez menor de
médicos.
Las supuestas etapas son las siguientes:
● Etapa 1: ya sea después de un evento o sin causa
aparente, el paciente desarrolla dolor en una extremidad. Las características
esenciales incluyen ardor, dolor punzante; dolor difuso e incómodo;
sensibilidad al tacto o al frío; y edema localizado [ 44 ]. La distribución del
dolor no es compatible con un solo nervio periférico, tronco o lesión de la
raíz. Las alteraciones vasomotoras ocurren con intensidad variable, produciendo
color y temperatura alterados. La radiografía generalmente es normal, pero puede
mostrar desmineralización irregular.
● Etapa 2: la segunda etapa está marcada por la
progresión del edema de los tejidos blandos, el engrosamiento de la piel y los
tejidos blandos articulares, el desgaste muscular y el desarrollo de la piel
endurecida. Esto puede durar de tres a seis meses.
● Etapa 3: la tercera etapa es más severa y se
caracteriza por la limitación del movimiento, el síndrome hombro-mano
(retracción capsular que produce un hombro congelado), contracturas de los
dedos, cambios céreos en la piel trófica y uñas quebradizas y quebradizas. La
radiografía ósea revela desmineralización severa.
EVALUACIÓN Y DIAGNÓSTICO
El diagnóstico de SDRC se
basa en las características clínicas determinadas por la historia y el examen
físico. El diagnóstico se sospecha en las siguientes circunstancias [ 32 ]:
● Los síntomas (p. Ej., Dolor, cambios sensoriales,
síntomas motores, disfunción autonómica o cambios tróficos) se desarrollan
después de un traumatismo de la extremidad, por lo general dentro de cuatro a
seis semanas.
● Los síntomas ya no se explican por completo por el
trauma inicial.
● Los síntomas afectan la extremidad distal, van más
allá de la región involucrada en el trauma o se extienden más allá del
territorio inervado por un solo nervio o raíz nerviosa.
El diagnóstico se puede hacer si los pacientes con
estos síntomas cumplen con los criterios de diagnóstico clínico para el SDRC
discutidos en la siguiente sección a continuación.
No existe una prueba o método "estándar de
oro" para confirmar el diagnóstico. Sin embargo, algunos investigadores
afirman que ciertas investigaciones son útiles para el diagnóstico de CRPS, en
particular la gammagrafía ósea en tres fases que muestra una mayor captación
del radiotrazador en las articulaciones distantes del sitio del traumatismo.
Otras pruebas con alguna utilidad posible incluyen radiografías lado a lado (p.
Ej., Ambas manos representadas en la misma radiografía) que muestran
descalcificación ósea irregular y mediciones repetidas a largo plazo de la
temperatura de la piel que muestran una diferencia> 1 ° C para el lado
afectado o no [ 32 ].
Los criterios de diagnóstico - Los criterios de
consenso de Budapest para el diagnóstico clínico de SDRC son las siguientes [
1,3 ]:
● Dolor continuo, que es desproporcionado a
cualquier evento incitador
● Para el diagnóstico clínico de SDRC, el paciente
debe informar al menos un síntoma en tres de las siguientes cuatro categorías
(los criterios de investigación más estrictos requieren al menos un síntoma en
las cuatro categorías):
• Sensorial: informes de hiperestesia y / o alodinia
• Vasomotor: informes de asimetría de temperatura y
/ o cambios en el color de la piel y / o asimetría del color de la piel
• Sudomotor / edema: informes de edema y / o cambios
de sudoración y / o sudoración por sudoración
• Motor / trófico: informes de disminución del rango
de movimiento y / o disfunción motora (debilidad, temblor, distonía) y / o
cambios tróficos (cabello, uñas, piel)
● Para el diagnóstico clínico de CRPS, el paciente
debe mostrar al menos un signo en el momento de la evaluación en dos de las
cuatro categorías siguientes (los criterios de investigación más estrictos
requieren al menos un signo en tres de las cuatro categorías):
• Sensorial: evidencia de hiperalgesia (al pinchazo)
y / o alodinia (a tacto ligero y / o sensación de temperatura y / o presión
somática profunda y / o movimiento articular)
• Vasomotor: evidencia de asimetría de temperatura
(> 1 ° C) y / o cambios en el color de la piel y / o asimetría
• Sudomotor / edema: evidencia de edema y / o
cambios en la sudoración y / o sudoración por sudoración
• Motor / trófico: evidencia de disminución del
rango de movimiento y / o disfunción motora (debilidad, temblor, distonía) y /
o cambios tróficos (cabello, uñas, piel)
● No hay otro diagnóstico que explique mejor los
signos y síntomas
Los criterios de Budapest no incorporan
explícitamente ciertas características clave del SDRC, incluida la afectación
de las extremidades, pero no la cabeza o el tronco, la aparición predominante
de síntomas y signos en la parte distal de la extremidad y la presencia de
síntomas y signos que no corresponden al territorio de un nervio o raíz
nerviosa [ 32 ]. Sin embargo, estos conceptos están encapsulados en la
definición de CRPS proporcionada por la conferencia del grupo de consenso de
Budapest [ 1 ] como se señaló anteriormente. (Ver 'Definición y terminología'
arriba).
Los criterios de Budapest parecen ser igualmente
sensibles y más específicos en comparación con los criterios de consenso de CRPS
anteriores [ 2,45 ] para diferenciar a los pacientes con SDRC de pacientes con
otros tipos de dolor neuropático [ 46 ].
La gammagrafía ósea - La gammagrafía ósea (es decir,
la gammagrafía ósea) es una técnica sensible para la detección de cualquiera de
una gran variedad de huesos, articulaciones y trastornos periarticulares,
incluyendo fractura, infección, tumor, artritis, y enfermedad ósea metabólica.
La gammagrafía ósea de triple fase es útil para detectar alteraciones en el
metabolismo óseo en pacientes con CRPS que tienen resorción ósea activa. Aunque
el CRPS es un diagnóstico clínico que no depende de los resultados de los
estudios por imágenes, la gammagrafía ósea realizada dentro de los primeros
cinco meses después del inicio de los síntomas puede respaldar el diagnóstico
si hay un aumento cuantitativo (ipsolateral> 1,32 en comparación con
contralateral) del radiotrazador evaluación de la región de interés durante la
fase de mineralización (es decir, tercera) en las articulaciones distantes del
sitio del trauma [32,47 ]. Es de destacar que una gammagrafía ósea negativa no
descarta el diagnóstico de SDRC [ 32 ].
La radiografía simple - radiografías simples con
frecuencia demuestran osteoporosis irregular, pero la sensibilidad de este
hallazgo para CRPS es muy baja [ 32 ]. Algunos expertos sugieren que la
obtención de imágenes con ambas manos en la misma radiografía puede ser útil
para el diagnóstico de SDRC si existe osteoporosis en los huesos que afecta la
mano afectada [ 32 ].
Pruebas autonómicas - autonómica pruebas que se han
utilizado para evaluar a los pacientes con sospecha de CRPS incluyen la salida
de descanso sudor (RSO), la temperatura de la piel en reposo (RST), y la prueba
del reflejo cuantitativo sudomotora axón (QSART). Algunos expertos abogan por
la medición en serie de las temperaturas de la piel [ 32 ], según la evidencia
de un estudio pequeño de que una diferencia de 2 ° C para el lado afectado
versus el lado no afectado apoyaba el diagnóstico de SDRC [ 48 ]. Sin embargo,
este método requiere un monitoreo de cinco a ocho horas con el registro de la
temperatura de la piel en intervalos de un minuto usando sensores de
temperatura aplicados a los dedos índice. Por lo tanto, no es práctico como una
prueba clínica de rutina [ 49 ].
Otras pruebas e intervenciones : no hay un papel
claro para la resonancia magnética (MRI) o la tomografía computarizada (CT) en
la evaluación de sospecha de SDRC, ni existe ningún papel para la respuesta a
la simpaticólisis para confirmar el diagnóstico de SDRC.
La RM puede ser útil para excluir algunas afecciones
en el diagnóstico diferencial (ver "Diagnóstico diferencial" a
continuación) pero no es útil para confirmar el diagnóstico de SDRC [ 32 ].
Datos limitados sugieren que la tomografía computarizada puede mostrar áreas
focales de osteoporosis en una apariencia de queso suizo [ 50 ]. Sin embargo,
sopesando el costo, la dosis de radiación y la experiencia limitada con el uso
de la tomografía computarizada en la evaluación de pacientes con SDRC,
sugerimos no utilizar la TC como prueba diagnóstica.
Históricamente, se consideró necesario el alivio
transitorio y abrupto del dolor y la disestesia con una simpatólisis química
sistémica (es decir, anestesia regional intravenosa, también denominada
"bloqueo de Bier" y / o bloqueo nervioso simpático regional como
ganglio estrellado o bloqueo neuromuscular simpático lumbar). para hacer el
diagnóstico de CRPS. Sin embargo, como el papel del sistema nervioso simpático
en la patogenia del SDRC sigue siendo poco claro y contradictorio, ahora se
acepta ampliamente que una respuesta positiva al bloqueo simpático no es un
diagnóstico de SDRC. Por el contrario, tal respuesta es un indicador importante
del dolor mantenido con simpatía.
DIAGNÓSTICO DIFERENCIAL
Las afecciones que pueden
presentar algunas de las características clínicas del SDRC incluyen las
siguientes [ 51 ]:
● Infección de la piel, los músculos, las
articulaciones o los huesos, que se caracteriza por enrojecimiento (eritema),
hinchazón (edema), calor y dolor. Las pruebas de laboratorio que son
indicadores de una infección, como aumentos en la velocidad de sedimentación
globular (ESR) o en la proteína C-reactiva (CRP) y las elevaciones en el
recuento de glóbulos blancos en la sangre periférica, son comunes en el SDRC
(Consulte "Análisis del líquido sinovial" y "Tenosinovitis
infecciosa" y "Descripción general de la osteomielitis en
adultos" y "Celulitis y absceso cutáneo: manifestaciones clínicas y
diagnóstico" ).
● Síndrome compartimental, causado por un aumento de
la presión dentro de un compartimento delimitado por membranas fasciales
inflexibles. El síndrome de compartimiento agudo es una emergencia quirúrgica
que generalmente se desarrolla poco después de un traumatismo significativo,
particularmente en las fracturas de huesos largos de la parte inferior de la
pierna o el antebrazo. El síndrome de compartimiento agudo también puede
ocurrir después de un traumatismo menor o por causas no traumáticas como lesión
por isquemia-reperfusión, coagulopatía, ciertos envenenamientos y mordeduras de
animales, extravasación de líquidos por vía intravenosa, inyección de drogas
recreativas y compresión prolongada de las extremidades. Los primeros síntomas
incluyen dolor progresivo fuera de proporción a la lesión; los signos incluyen
compartimientos hinchados y dolorosos con estiramiento pasivo de los músculos
dentro del compartimento afectado.
● Enfermedad vascular periférica, que provoca
hipoperfusión de los pies y las piernas, lo que provoca pies fríos con
decoloración, calambres en las piernas o dolor que empeora con la actividad y
mejora con el reposo (es decir, claudicación vascular). (Consulte
"Características clínicas y diagnóstico de la enfermedad arterial
periférica de la extremidad inferior" ).
● Trombosis venosa profunda, que puede provocar
hinchazón, enrojecimiento y dolor en las extremidades involucradas. Sin embargo,
este problema en general se puede distinguir de CRPS por la historia y el
examen físico junto con la prueba vascular de ultrasonido Doppler. (Consulte
"Presentación clínica y diagnóstico del adulto no gestante con sospecha de
trombosis venosa profunda de la extremidad inferior" ).
● Neuropatía periférica (p. Ej., Neuropatía
diabética), que ocasionalmente puede presentarse con hipersensibilidad y
cambios distróficos de las extremidades.
● Síndrome de salida torácica vascular, que puede
presentarse con hinchazón (a menudo una sensación de hinchazón sin cambio
visible), parestesia y eritema.
● La artritis reumatoide, una enfermedad autoinmune
inflamatoria crónica que afecta la sinovia de las articulaciones, puede tener
signos y síntomas similares a los del CRPS. Sin embargo, los síntomas de la
artritis reumatoide están presentes en varias articulaciones (p. Ej., Muñecas,
rodillas, hombros, manos y pies), a diferencia de solo una región del cuerpo.
Además, el análisis de sangre de laboratorio es útil para diagnosticar la
artritis reumatoide. (Consulte "Diagnóstico y diagnóstico diferencial de
la artritis reumatoide" ).
● Fenómeno de Raynaud, una respuesta
vasoconstrictora exagerada a la temperatura fría y al estrés emocional. El
fenómeno se manifiesta clínicamente por cambios de color claramente demarcados
de la piel de los dedos. El diagnóstico del fenómeno de Raynaud se realiza si
los dedos son inusualmente sensibles al frío y cambian de color (a blanco, azul
o ambos) cuando se exponen a temperaturas frías. Se cree que la
vasoconstricción anormal de arterias digitales y arteriolas cutáneas debido a
un defecto local en las respuestas vasculares normales subyace al trastorno.
(Consulte "Manifestaciones clínicas y diagnóstico del fenómeno de Raynaud"
).
● Erythromelalgia, un síndrome clínico adquirido o
(raramente) hereditario caracterizado por la aparición intermitente de
extremidades rojas, calientes y dolorosas. El síndrome generalmente afecta las
piernas (predominantemente los pies) pero también puede involucrar brazos
(predominantemente manos) y rara vez afecta a la cara. (Ver
"Erythromelalgia" ).
● Trastorno de conversión, una afección involuntaria
en la que los síntomas neurológicos están presentes en ausencia de una
enfermedad neurológica pero no son fingidos. (Consulte "Trastorno de
conversión en adultos: terminología, diagnóstico y diagnóstico
diferencial" ).
● Trastorno facticio, una producción intencional de
síntomas o hallazgos físicos o psicológicos para asumir el "papel de
enfermo". (Ver "trastorno facticio impuesto al yo (síndrome de
Munchausen)" ).
REFERENCIAS
Harden RN,
Bruehl S, Stanton-Hicks M, Wilson PR. Nuevos criterios de
diagnóstico propuestos para el síndrome de dolor regional complejo. Pain Med 2007; 8: 326.
Stanton-Hicks M,
Jänig W, Hassenbusch S, y col. Distrofia simpática refleja:
conceptos cambiantes y taxonomía. Pain 1995; 63: 127.
Harden RN, Oaklander AL, Burton AW, y col. Síndrome
de dolor regional complejo: pautas prácticas de diagnóstico y tratamiento, 4ª
edición. Pain Med 2013; 14: 180.
Bruehl S. Una actualización sobre la fisiopatología
del síndrome de dolor regional complejo. Anestesiología 2010; 113: 713.
Oaklander AL, Fields HL. ¿Es la distrofia simpática
refleja / síndrome de dolor regional complejo tipo I una neuropatía de fibras
pequeñas? Ann Neurol 2009; 65: 629.
Eberle T, Doganci B, Krämer HH, y col. Síndromes de
dolor regionales complejos cálidos y fríos: ¿diferencias más allá de la
temperatura de la piel? Neurology 2009; 72: 505.
Bussa M, Guttilla D, Lucia M, et al. Síndrome de
dolor regional complejo tipo I: una revisión exhaustiva. Acta Anaesthesiol
Scand 2015; 59: 685.
Munnikes RJ, Muis C, Boersma M, y col. El síndrome
de dolor regional complejo de etapa intermedia tipo 1 no está relacionado con
las citocinas proinflamatorias. Mediators Inflamm 2005; 2005: 366.
Alexander GM, van Rijn MA, van Hilten JJ, y otros
Cambios en los niveles de líquido cefalorraquídeo o citoquinas proinflamatorias
en CRPS. Pain 2005; 116: 213.
Uçeyler N, Eberle T, Rolke R, y col. Patrones de
expresión diferencial de citoquinas en el síndrome de dolor regional complejo.
Pain 2007; 132: 195.
Parkitny L, McAuley JH, Di Pietro F, y col.
Inflamación en el síndrome de dolor regional complejo: una revisión sistemática
y metaanálisis. Neurology 2013; 80: 106.
Marinus J, Moseley GL, Birklein F, y col.
Características clínicas y fisiopatología del síndrome de dolor regional
complejo. Lancet Neurol 2011; 10: 637.
Jänig W, Baron R. Síndrome de dolor regional
complejo: ¿misterio explicado? Lancet
Neurol 2003; 2: 687.
Wasner G,
Schattschneider J, Heckmann K, y col. Anomalías vasculares en
la distrofia simpática refleja (CRPS I): mecanismos y valor diagnóstico. Brain
2001; 124: 587.
Maihöfner C, Handwerker HO, Neundörfer B, Birklein
F. Reorganización cortical durante la recuperación del síndrome de dolor
regional complejo. Neurology 2004; 63: 693.
Pleger B, Tegenthoff M, Ragert P, y col. La
resintonización sensoriomotora [corregida] en el síndrome de dolor regional
complejo es paralela a la reducción del dolor. Ann Neurol 2005; 57: 425.
Freund W,
Wunderlich AP, Stuber G, y col. Diferente activación de
la corteza opercular y cingulada posterior (PCC) en pacientes con síndrome de
dolor regional complejo (CRPS I) en comparación con controles sanos durante la
percepción del dolor inducido eléctricamente: un estudio de resonancia
magnética funcional. Clin
J Pain 2010; 26: 339.
Maihöfner C,
Baron R, DeCol R, y col. El sistema motor muestra cambios
adaptativos en el síndrome de dolor regional complejo. Brain 2007; 130: 2671.
Kemler MA, el AC Vusse, van den Berg-Loonen EM, et
al., HLA-DQ1 asociado con distrofia simpática refleja. Neurology 1999; 53:
1350.
van de Beek WJ, Roep BO, van der Slik AR, y col. Los
loci de susceptibilidad para el síndrome de dolor regional complejo. Pain 2003;
103: 93.
Allen G, Galer BS, Schwartz L. Epidemiología del
síndrome de dolor regional complejo: una revisión retrospectiva de 134
pacientes. Pain 1999; 80: 539.
Sandroni P, Benrud-Larson LM, McClelland RL, PA
baja. Síndrome de dolor regional complejo tipo I: incidencia y prevalencia en
el condado de Olmsted, un estudio basado en la población. Pain 2003; 103: 199.
de Mos M, de Bruijn AG, Huygen FJ, et al. La
incidencia del síndrome de dolor regional complejo: un estudio basado en la
población. Pain 2007; 129: 12.
Schwartzman RJ, Erwin KL, Alexander GM. La historia
natural del síndrome de dolor regional complejo. Clin J Pain 2009; 25: 273.
Pak TJ, Martin
GM, Magness JL, Kavanaugh GJ. Distrofia simpática refleja.
Revisión de 140 casos. Minn
Med 1970; 53: 507.
Geertzen JH, de
Bruijn H, de Bruijn-Kofman AT, Arendzen JH. Distrofia simpática
refleja: tratamiento temprano y aspectos psicológicos. Arch Phys Med Rehabil
1994; 75: 442.
Birklein F, Riedl B, Sieweke N, y col. Hallazgos
neurológicos en síndromes de dolor regional complejo - análisis de 145 casos.
Acta Neurol Scand 2000; 101: 262.
Bruehl S, Carlson CR. Factores psicológicos predisponentes
en el desarrollo de la distrofia simpática refleja. Una revisión de la
evidencia empírica. Clin J Pain 1992; 8: 287.
Beerthuizen A, van 't Spijker A, Huygen FJ, et al.
¿Existe una asociación entre los factores psicológicos y el síndrome de dolor
regional complejo tipo 1 (CRPS1) en adultos? Una revisión sistemática Pain
2009; 145: 52.
Beerthuizen A, Stronks DL, Huygen FJ, et al. La
asociación entre los factores psicológicos y el desarrollo del síndrome de
dolor regional complejo tipo 1 (CRPS1) - un estudio prospectivo multicéntrico.
Eur J Pain 2011; 15: 971.
Lohnberg JA, Altmaier EM. Una revisión de los
factores psicosociales en el síndrome de dolor regional complejo. J Clin
Psychol Med Settings
2013; 20: 247.
Birklein F, O'Neill D, Schlereth T. Síndrome de
dolor regional complejo: una perspectiva optimista. Neurology 2015; 84:89.
Thomson McBride
AR, Barnett AJ, Livingstone JA, Atkins RM. Síndrome de dolor
regional complejo (tipo 1): una comparación de 2 métodos de criterios de
diagnóstico. Clin J Pain 2008; 24: 637.
Barrera P, van Riel PL, de Jong AJ, et al. Síndrome
de distrofia simpática refleja recurrente y migratoria. Clin Rheumatol 1992; 11: 416.
Maleki J, LeBel
AA, Bennett GJ, Schwartzman RJ. Patrones de
diseminación en el síndrome de dolor regional complejo, tipo I (distrofia
simpática refleja). Pain 2000; 88: 259.
van Rijn MA, Marinus J, Putter H, y col. Difusión
del síndrome de dolor regional complejo: no es un proceso aleatorio. J Neural
Transm (Viena) 2011; 118: 1301.
Homans J. causalgia menor: un síndrome neurovascular
hiperestésico. N Engl J Med 1940; 222: 870.
DEJONG R, CULLEN SC. ASPECTOS TEÓRICOS DEL DOLOR:
FENÓMENOS DEL DOLOR BIZARRE DURANTE LA ANESTESIA ESPINAL BAJA. Anestesiología
1963; 24: 628.
Bonica JJ. Causalgia y otras distrofias simpáticas
reflejas. Postgrad Med 1973; 53: 143.
STEINBROCKER O, ARGYROS TG. El síndrome hombro-mano:
estado presente como entidad diagnóstica y terapéutica. Med Clin North Am 1958;
42: 1533.
Mowat AG. Tratamiento del síndrome hombro-mano con
corticosteroides. Ann Rheum
Dis 1974; 33: 120.
Veldman PH,
Reynen HM, Arntz IE, Goris RJ. Signos y síntomas de la distrofia
simpática refleja: estudio prospectivo de 829 pacientes. Lancet 1993; 342:
1012.
Freedman M, Greis AC, Marino L, y col. Síndrome de
dolor regional complejo: diagnóstico y tratamiento. Phys Med Rehabil Clin N Am 2014; 25: 291.
Sheon RP,
Moskowitz RW, Goldberg VM. Dolor reumático de tejido blando:
reconocimiento, manejo, prevención, tercera edición, Williams & Wilkins,
Baltimore 1996. p.116.
Clasificación de dolor crónico, 2nd ed., Mersky H,
Bogduk N (Eds), IASP Press, Seattle 1994.
Harden RN, Bruehl S, Pérez RS, y col. Validación de
los criterios de diagnóstico propuestos (los "Criterios de Budapest")
para el síndrome de dolor regional complejo. Pain 2010; 150: 268.
Wüppenhorst N, Maier C, Frettlöh J, y col.
Sensibilidad y especificidad de la gammagrafía ósea de 3 fases en el
diagnóstico del síndrome de dolor regional complejo de la extremidad superior. Clin J Pain 2010; 26: 182.
Krumova EK,
Frettlöh J, Klauenberg S, et al. Mediciones de
temperatura de la piel a largo plazo: una herramienta de diagnóstico práctica
en el síndrome de dolor regional complejo. Pain 2008; 140: 8.
Cohen SP, Raja SN. ¿La medición prolongada de la
temperatura de la piel mejora el diagnóstico del síndrome de dolor regional
complejo? Nat Clin Pract Neurol 2009; 5:14.
Sambrook P, campeón GD. Distrofia simpática refleja;
cambios característicos en el hueso en la tomografía computarizada. J Rheumatol
1990; 17: 1425.
Turner-Stokes L, Goebel A, Grupo de desarrollo de
directrices. Síndrome de dolor regional complejo en adultos: orientación
concisa. Clin Med (Lond







































