Un paciente de sexo masculino de 20 años de edad
consultó por hinchazón en pantorrilla izquierda y púrpura petequial en piernas.
Su
enfermedad actual había comenzado 20 días antes con odinofagia, tos seca, resfrío y escalofríos. En ese momento notó que
tenía costras en las fosas nasales. A su hermano, con un cuadro similar, se le
diagnosticó faringitis estreptocócica unos días antes. El paciente había estado acampando en una
región del noroeste de los EEUU, al regresar del cual presentó casi inmediatamente calambre e hinchazón en
la pierna izquierda. Se llevó a cabo una ecografía y eco Doppler de la pierna
sospechando TVP la cual fue normal. Fue medicado con ibuprofeno y azitromicina
sospechando infección de vías aéreas. El dolor lejos de mejorar empeoró
agregándose un rash eritematoso en el dorso de ambos pies.
El
paciente tenía antecedentes de enfermedad de Crohn desde los 10 años de edad
por lo que recibió corticoterapia prolongada hasta hacía tres años cuando
comenzó con adalimumab semanal que le permitió reducir la dosis de corticoides
y permitió que el paciente tuviese un marcado mejoramiento de su desarrollo ya que ganó en peso y altura con
el cambio de tratamiento. Los años previos el paciente había tenido
varios episodios autolimitados de dolor
de garganta, rinorrea y costras nasales con sangrados asociados para lo que
tomaba ibuprofeno. No fumaba ni tomaba alcohol. Su hermana tenía también
enfermedad de Crohn.
Estaba
afebril, TA 120/70 mmHg, sat O2 97%
respirando aire ambiente. Se quejaba de dolor en la pierna. Las costras nasales
habían mejorado y no tenía úlceras ni perforación de tabique ( Figura 1)
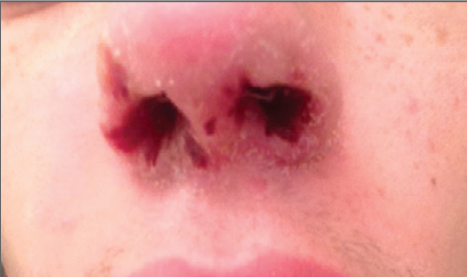 |
| Tres semanas antes del cuadro de presentación presentó costras en ambas narinas. |
El
examen cardiovascular y respiratorio eran normales. Había dolor a la palpación
de la pantorrilla izquierda y el dorso del pie sin eritema, calor ni signo de
la fóvea. Había sinovitis en el tobillo izquierdo. Los pulsos normales. Había
lesiones purpúricas no dolorosas, algunas apenas palpables en ambas piernas
pero especialmente en la izquierda (Figura 2B).
Había dos nódulos dolorosos de 1 cm de diámetro cada uno en la región
supramaleolar interna derecha (Figura 2C) y eran similares a lesiones que
habían salido en el pasado.
 |
| Figura 2A |
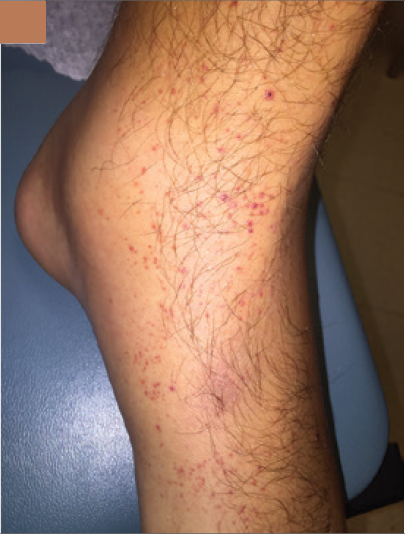 |
| Figura 2B |
 |
| Fig 2C |
Figura 2 En la presentación había inflamación y dolor de la pierna izquierda a nivel desde la pantorrilla hasta la región dorsal del pie (2A). Una púrpura palpable estaba presente en ambos pies (2B). Dos nódulos dolorosos de 1 cm cada uno con eritema estaban presentes en maléolo interno (2C)
Los
electrolitos séricos, glucosa, creatin kinasa, C3 y C4 eran normales, como
también eran normales la función renal y hepática, el hemograma y el recuento
diferencial.
Los test
de FAN, ANCA eran negativos. El antígeno de superficie para hepatitis B, los
anticuerpos para hepatitis C, HIV tipo 1, y 2, y antígeno p24 eran negativos. Los hemocultivos fueron
negativos. La orina era amarillo clara con una densidad de 1014 y un pH de 5,5,
sin glucosa, cetonas ni bilirrubina, proteínas, sangre o nitratos en las tiras reactivas.
El sedimento no mostraba células rojas , blancas, bacterias ni cilindros. La
proteína C reactiva era de 2.0 mg/dl (normal 0 a 0.9), la VSG era de 30 mm/h y
las ASTO de 400 UI/ml (rango normal 0 a 200).
La Rx de
tórax mostró una silueta cardíaca normal
sin consolidaciones ni derrame pleural. La TC con contraste EV de tibia, peroné y pie mostró estructuras
óseas normales sin fracturas ni erosiones corticales así como edema difuso
inespecífico de partes blandas del tobillo y parte proximal del pie sin absceso
ni engrosamientos fasciales.
El
paciente fue internado y se llevaron a cabo durante su internación varios test
diagnósticos
Cuál es
el/los diagnósticos diferenciales que pueden plantearse en este caso?
DIAGNÓSTICO
DIFERENCIAL
Este
paciente de 20 años con antecedentes de enfermedad de Crohn estaba recibiendo tratamiento
con adalimumab se presentó con púrpura palpable, monoartritis con hinchazón
prominente, lesiones nodulares de piel y costras nasales en el contexto de enfermedad
de vía aérea superior. Estos hallazgos son poco frecuentes en un paciente
joven, y diseñar una lista de diagnósticos diferenciales puede ayudar a
estrechar la lista de posibilidades diagnósticas. Dado que la presencia de costras nasales y lesiones
nodulares el paciente las había presentado previamente en el pasado, habría que enfocar el diagnóstico
diferencial en las nuevas
manifestaciones de: púrpura palpable
asociada a monoartritis
PÚRPURA
Y ARTRITIS
Las
lesiones purpúricas son causadas generalmente por extravasación de sangre desde
vasos dañados. La púrpura no palpable puede ser debida a trombocitopenia, enfermedad
sistémica (por ej amiloidosis),
infecciones, estados hipercoagulables, deficiencias nutricionales (por ej
escorbuto), exposición solar (por ej púrpura solar), y entidades idiopáticas
(por ej enfermedad de Schamberg).1 Sin
embargo, la púrpura palpable que involucra
áreas declives del cuerpo, sometidas a
mayor ley de gravedad como se vio en este paciente, es muy sugerente de una
vasculitis de pequeños vasos. La presencia de artritis inflamatoria en este paciente es una
evidencia adicional de que un proceso
vasculítico es el que está causando la púrpura; la artritis se ve comúnmente en
pacientes con vasculitis pero serían poco usuales en pacientes con otras causas de púrpura.
VASCULITIS
Cuando
evalúo por primera vez a un paciente con
vasculitis, intento determinar si los
vasos involucrados son pequeños, medianos o grandes. Esta categorización de las
vasculitis sistémicas ofrece un marco diagnóstico útil, porque cada tamaño de
vaso afectado está asociado con características clínicas características (Tabla
1). Por ejemplo, la vasculitis de vasos grandes típicamente afecta la aorta y
sus ramas y no causa lesiones de
tipo purpúricas en piel como las que se ven en este paciente Dado a que las
arterias de tamaño mediano suministran vascularización a áreas más grandes de la piel que las arterias
pequeñas, las vasculitis de vasos medianos producen úlceras, nódulos y livedo racemosa en la piel,
mientras que las vasculitis de pequeños
vasos típicamente causan petequias y púrpura.5
La manifestación de la piel predominante
de este paciente es una púrpura palpable, lo que sugiere que tiene una vasculitis que afecta las pequeñas
arterias.

TABLA 1
ESPECTRO DE LAS VASCULITIS DE VASOS GRANDES MEDIANOS Y PEQUEÑOS
Después
de evaluar el tamaño de los vasos afectados, verifico el grado de participación
de otros órganos, lo que determina la urgencia de la evaluación y la severidad
del proceso patológico. Este paciente no tiene proteinuria o hematuria, lo
cual sugeriría glomerulonefritis, y no
tiene hemoptisis o infiltrados parenquimatosos pulmonares, lo cual sugeriría hemorragia alveolar difusa. Además,
tampoco tiene síntomas neuropáticos, lo cual sugeriría una neuropatía vasculítica y no
tiene síntomas abdominales lo que podrían sugerir vasculitis involucrando los vasos mesentéricos.
VASCULITIS
DE PEQUEÑOS VASOS
Las
vasculitis de pequeños vasos comparten muchas características clínicas
comunes. Por ejemplo, síntomas constitucionales, artritis inflamatoria, púrpura
palpable, glomerulonefritis, hemorragia alveolar difusa, y neuropatía vasculítica
puede ocurrir en casi todas las vasculitis de pequeños vasos. Para evaluar
causas potenciales de una vasculitis de pequeño vaso, me centro
en las características clínicas que pueden ayudar a diferenciar entre las
vasculitis (Tabla 1).
Vasculitis
secundarias
Una de
las características más notables de la presentación de este paciente es el
hallazgo de las lesiones en la piel. Las vasculitis cutáneas pueden ocurrir
como manifestación de cáncer, infección o una condición inflamatoria.6 Antes de
considerar las muchas vasculitis
sistémicas primarias, es importante determinar si la vasculitis en este
paciente puede deberse a alguno de esos procesos mencionados. El cáncer es poco probable
en este joven paciente que no tiene síntomas constitucionales, linfadenopatías,
o una masa testicular. Dado a que este
paciente está recibiendo un inhibidor del factor necrosis tumoral alfa (TNF-α),
tiene riesgo aumentado de infección.7
Además, su reciente viaje de campamento al noroeste de los Estados Unidos puede
haberlo puesto en riesgo de una enfermedad transmitida por garrapatas, como la
fiebre manchadade las Montañas Rocosas. Sin embargo, infecciones que causan
púrpura y petequias, tales como la fiebre manchada de las Montañas Rocosas y la
meningococcemia diseminada, son típicamente graves y de inicio repentino, y
estas características no se ajustan con la presentación de este paciente.8
Infecciones como como la endocarditis bacteriana subaguda puede causar vasculitis
cutánea a través de la formación de complejos antígeno-anticuerpo. Sin embargo,
esto usualmente ocurre con un curso de
tiempo gradual, y el recuento normal de glóbulos blancos y la ausencia de síntomas
y factores de riesgo de endocarditis en este paciente hacen que este
diagnóstico sea improbable. El uso de drogas ilícitas, como la cocaína
(particularmente en combinación con levamisol, un elemento común adulterante de
la cocaína), puede causar vasculitis. Sin embargo, este paciente informa que no
tiene antecedentes de uso de cocaína, y sus lesiones no son típicas de las
vasculitis inducidas por levamisol, que
afecta clásicamente los pabellones auriculares. La vasculitis también puede ser
el resultado de condiciones inflamatorias tales como el lupus eritematoso sistémico y el síndrome
de Sjögren. Este paciente no tiene síntomas que sugieran la presencia de
ninguna de estas estas condiciones, y la prueba negativa para anticuerpos antinucleares
(FAN), hace que estos diagnósticos sean muy improbables. La enfermedad de Crohn
tiene una serie de manifestaciones extraintestinales, y en raras ocasiones, se
ha informado vasculitis concomitante. Sin embargo, la mayoría de los casos
reportados son de vasculitis primarias en asociación con Enfermedad de Crohn.10
No puedo descartar la posibilidad de vasculitis relacionada con la enfermedad
de Crohn, pero dado su rareza, consideraría que una vasculitis sistémica
primaria es la causa más probable del cuadro de este paciente
Vasculitis
sistémica primaria
Las
vasculitis sistémicas de pequeños
vasos se pueden dividir en dos categorías:
vasculitis asociada a ANCA y vasculitis
mediada por complejos inmunes. Hay tres tipos de vasculitis asociadas a ANCA: la granulomatosis eosinofílica con
poliangeítis (Churg-Strauss), la
poliangeítis microscópica, y la granulomatosis con poliangeítis (enfermedad de
Wegener). En este paciente, la ausencia
de eosinofilia periférica descarta la granulomatosis eosinofílica con
poliangeitis La prueba negativa para ANCA combinada con la ausencia de enfermedad
renal y pulmonar hace que la poliangeítis microscópica sea una consideración
diagnóstica poco probable. Sin embargo, la
granulomatosis con poliangeítis debe ser considerada, porque puede causar
artritis, vasculitis cutánea y lesiones inflamatorias granulomatosas en la
nariz, senos paranasales, pulmones y vías respiratorias.2 Las costras nasales
son una manifestación común de granulomatosis con poliangeítis con afectación
nasosinusal, y es una característica importante que distingue a la
granulomatosis con poliangeítis de las sinusitis viral o bacteriana, que
típicamente no causan costras. En pacientes que tienen granulomatosis con
poliangeítis, la sensibilidad de las pruebas para ANCA es más del 90% entre
aquellos que tienen participación renal,
pero es aproximadamente del 60% entre aquellos que no tienen afectación
renal2,3. Aunque una prueba negativa para ANCA no descarta granulomatosis con poliangeítis en este caso,
yo no hubiese esperado que las costras nasales se resolvieran espontáneamente
en un paciente con granulomatosis y
poliangeítis como aquí sucedió. En ausencia de persistencia manifestaciones
granulomatosas, una prueba negativa para ANCA es tranquilizador y hace que la
granulomatosis con poliangeítis sea un diagnóstico poco probable.
Vasculitis
mediada por complejos inmunes
Aparte
de vasculitis asociada con reacciones a medicamentos, el tipo más común de vasculitis mediada por complejos inmunes son la
vasculitis crioglobulinémica y las vasculitis por
IgA (anteriormente Púrpura de Schönlein-Henoch). La vasculitis
crioglobulinémica puede ser idiopática pero se ve típicamente en asociación con
infección por el virus de la hepatitis C y es menos comúnmente vista con lupus
eritematoso sistémico, síndrome de Sjögren o infección por el virus de la
hepatitis B. 11 La vasculitis crioglobulinémica es el resultado de la
deposición del complejo inmune en las paredes de los vasos, lo que conduce a la
activación de la vía clásica del complemento. Como resultado, el nivel de C4 en
sangre es bajo en la mayoría de los pacientes afectados. A pesar de que los
niveles de crioglobulina no fueron controlados en este paciente, la prueba
negativa para el virus de la hepatitis C y los niveles normales de complemento
hacen de la vasculitis crioglobulinémica un diagnóstico poco probable. La presencia de
púrpura palpable en este paciente obliga a la consideración de vasculitis por IgA porque, a diferencia de
otras vasculitis, la vasculitis por IgA
se asocia con lesiones de inicio cutáneo antes o poco después del inicio de las
manifestaciones extracutáneas en la mayoría de los casos12. Aunque la
vasculitis IgA ocurre con mayor frecuencia en niños que en adultos, es una
vasculitis común en adultos, con una incidencia mayor que la de granulomatosis
con poliangitis.13,14 Este paciente no tuvo afectación gastrointestinal ni
renal, ambos se ven comúnmente en pacientes con vasculitis por IgA. Sin
embargo, aproximadamente un tercio de los pacientes con vasculitis por IgA no tiene afectación renal o
gastrointestinal.12,15 Aunque las manifestaciones articulares de la vasculitis por
IgA pueden involucrar cualquier
articulación, tienden a involucrar las grandes articulaciones de las piernas.
Prominente hinchazón de la pierna ha sido reportado con vasculitis por IgA.16
En resumen, la combinación de púrpura palpable y artritis con hinchazón
prominente de la pierna que se vio en este paciente es consistente con un
diagnóstico de vasculitis por IgA La vasculitis por IgA es única entre las
vasculitis porque con frecuencia es provocada por una infección o por uso de
medicamentos y con frecuencia es autolimitada.12 Un número de desencadenantes potenciales están
presentes en este paciente, incluida una infección respiratoria superior
reciente y el uso de un inhibidor de TNF-α. No está claro si la infección
respiratoria superior que precedió a la vasculitis de pequeños vasos en este
paciente fue causado por estreptococos del grupo A, que según los informes, el
hermano del paciente había tenido recientemente. Un solo título elevado de antiestreptolisina O podría
reflejar una infección ocurrida en el pasado remoto, 17 mientras que la
evidencia de una elevación de los títulos de estreptolisina O sería más sugestivo de una infección reciente.
Sin embargo, una amplia gama de infecciones, incluida la estreptocócica y las infecciones no estreptocócicas, han
estado involucradas en el desarrollo de vasculitis por IgA.18 Además, este paciente estaba recibiendo
un inhibidor del TNF-α, y algunos informes plantean la
posibilidad que esta clase de medicamentos puede conducir al desarrollo de
vasculitis por IgA.19 Características de presentación de este paciente que serían
atípicos en un paciente con vasculitis por IgA incluyen las lesiones nodulares y la
costra nasal. El hecho de que él había tenido estos síntomas intermitentemente antes
de que comience esta enfermedad sugiere que pueden no estar relacionados con el
corriente episodio. Aunque su causa sigue siendo incierta, la enfermedad de
Crohn subyacente del paciente ofrece una explicación plausible para ambos
síntomas. Las manifestaciones cutáneas de la enfermedad de Crohn incluyen
pioderma gangrenoso, dermatosis neutrofílicas y eritema nodoso.20 A pesar del
hecho de que las manifestaciones intestinales de este paciente parecen estar
bien controladas, sus lesiones nodulares pueden ser compatibles con eritema
nodoso. En raras ocasiones, la formación de costras nasales ha sido informado
como una manifestación extraintestinal de Enfermedad de Crohn, 21 y por lo
tanto podría ser evidencia de que tiene una enfermedad de Crohn activa, aunque otras
causas de formación de costras nasales
también son posibles.
En
resumen, a falta de signos y síntomas de cáncer e infección, una vasculitis
primaria sistémica de pequeño vaso es la explicación más probable de las
características de presentación de este
paciente. Dado la presencia de púrpura palpable y artritis con prominente hinchazón
periarticular y la ausencia de características sugerentes de otras vasculitis,
la vasculitis por IgA es el diagnóstico más probable en este paciente. La presencia de varios
desencadenantes posibles de la vasculitis por IgA apoyan más apoya este
diagnóstico Sospecho que el test diagnóstico
fue una biopsia de piel. Una tinción con
hematoxilina y eosina de una muestra de biopsia revelaría una vasculitis
leucocitoclástica, que es una enfermedad típica de encontrar en la biopsia de
piel en un paciente con cualquier vasculitis de pequeños vasos y no es
específico para vasculitis por IgA. Para
distinguir entre vasculitis por IgA y otras causas de vasculitis
leucocitoclástica, la tinción de inmunofluorescencia se necesitaría una muestra
de biopsia para evaluar depósitos de IgA en las paredes del vaso.
DIAGNÓSTICO
PRESUNTIVO
VASCULITIS
POR IGA.
DISCUSIÓN
PATOLÓGICA
La evaluación
microscópica de un punch de biopsia por punción de 4 mm de piel lesionada dela
zona del tobillo izquierdo, con una
tinción con hematoxilina-eosina reveló un infiltrado inflamatorio de la dermis
media y superficial con inflamación perivascular e intersticial compuesta predominantemente de neutrófilos. Era
prominente la leucocitoclasia, polvo nuclear de neutrófilos, extravasación de
eritrocitos, y necrosis fibrinoide focal de las paredes de los vasos estaban presentes (Fig. 3). Estos hallazgos
fueron consistentes con el diagnóstico
de vasculitis leucocitoclástica.
Figura
3. Muestra de biopsia de piel del
tobillo izquierdo (hematoxilina y eosina).
Se
muestra un infiltrado inflamatorio perivascular superficial y medio dérmico
(Panel A). Está compuesto predominantemente de neutrófilos, y hay leucocitosis
prominente y extravasación de eritrocitos (Paneles B y C). Hay necrosis fibrinoide
focal de las paredes de los vasos (Panel
D, flecha). Estos hallazgos son consistentes con el diagnóstico de vasculitis leucocitoclástica.
La
vasculitis leucocitoclástica representa un patrón de reacción vascular en la piel y no es un
diagnóstico
específico. Puede ser una manifestación de vasculitis por IgA o vasculitis
urticarial o puede ser idiopática. También puede ser el resultado de
infecciones subyacentes, uso de medicamentos, trastornos inflamatorios y cáncer.25,26
La patogénesis implica el depósito de complejos inmunes circulantes en las
paredes de vasos pequeños y activación del sistema del complemento. La
afluencia posterior de neutrófilos conduce a la liberación de enzimas
lisosómicas, que dañan la pared del vaso, lo que lleva al depósito de fibrina y
extravasación de eritrocitos.27 Este es un proceso dinámico con variación en la
cantidad de depósitos de complejos inmunes, y la histopatología características
observadas en una muestra de biopsia que tenga entre 18 a 24 horas de antigüedad desde su aparición
tienen más probabilidades de conducir a un diagnóstico de certeza.28 Los
hallazgos histopatológicos característicos de las vasculitis leucocitoclásticas
incluye un infiltrado rico en
neutrófilos que rodea vénulas poscapilares y asas capilares con leucocitoclasia,
hinchazón del endotelio vascular, necrosis fibrinoide de las paredes de los
vasos y extravasación de eritrocitos.26-28 Para ayudar a reducir el diagnóstico
diferencial de las vasculitis
leucocitoclásticas, se puede realizar inmunofluorescencia directa. Muchos pacientes
con vasculitis leucocitoclástica tienen evidencia de inmunorreactividad
perivascular para IgG, IgM, C3 y fibrinógeno en la inmunofluorescencia directa.25,26,29 En este
caso, una segunda muestra de biopsia por punción de 4 mm de piel afectada
obtenida de la zona del tobillo izquierdo
mostró inmunorreactividad
perivascular granular positiva para IgA, C3 y fibrina, resultados consistentes
con vasculitis leucocitoclástica asociada a IgA (Fig. 4).
Figura
4. Muestra de biopsia de piel del tobillo izquierdo (Inmunofluorescencia
directa).
La
inmunorreactividad positiva para IgA (paneles A y B), C3 (panel C) y fibrina
(panel D) está presente en forma de patrón granular, perivascular. Este patrón de tinción
es consistente con el diagnóstico de vasculitis IgA.
Es
importante tener presente que La
vasculitis leucocitoclástica asociada a IgA no es patognomónico de vasculitis
IgA, ya que depósitos de IgA también se
puede ver en vasculitis leucocitoclásticas asociada a crioglobulinemia o al uso de ciertos
medicamentos, incluidos los inhibidores del TNF-α.30,31 Sin embargo, en este caso, el diagnóstico anatomopatológicode vasculitis por
IgA se confirmó considerando los hallazgos en el contexto de la presentación
clínica de este paciente
DISCUSIÓN
DEL MANEJO
La
vasculitis IgA ocurre con mayor frecuencia en niños, y la mayoría de los casos
demuestran ser enfermedades autolimitadas que no requieren terapia. Los cuatro
sistemas de órganos que están más comúnmente afectados por la enfermedad son: la
piel, las articulaciones, el intestino y los riñones. Cuando las
manifestaciones clínicas en cualquiera de estos órganos son suficientemente
severos, el tratamiento debe ser considerado no solo para el alivio de los
síntomas, sino también para prevenir daño orgánico relacionado con la vasculitis.
En este paciente, la enfermedad cutánea, que se limitaba a una pequeña porción
de la piel y que no se asociaba con ulceración
cutánea, fue relativamente leve y no requirió tratamiento. El paciente
no tenía síntomas gastrointestinales en
la presentación. La participación
gastrointestinal en la vasculitis IgA
suele ser caracterizada por dolor abdominal cólico que a menudo ocurre antes de
la erupción de la púrpura palpable en la
piel y, a veces conduce a una cirugía exploratoria por un presunto abdomen
agudo. El paciente tampoco tenía evidencia de glomerulonefritis, una
complicación de la vasculitis por IgA,
la cual es particularmente difícil de tratar y puede
desarrollarse incluso durante las semanas o meses posteriores a la presentación inicial. Sin embargo, sus
síntomas musculoesqueléticos fueron severos; el paciente era incapaz de caminar debido a su artritis de
tobillo y el intenso dolor e hinchazón de su músculo gastrocnemio (gemelo).
Dado la gravedad de los síntomas musculoesqueléticos, se optó por iniciar el
tratamiento con glucocorticoides. Los glucocorticoides son efectivos en el
tratamiento de la vasculitis por IgA.32
Debido a que el paciente había utilizado corticoides crónicamente por su enfermedad de Crohn, se pensó que era importante usar la dosis efectiva más
baja comenzando con un curso de prednisona de 20 mg por día y la consigna de
reducir rápidamente la dosis. La púrpura del paciente se resolvió rápidamente y
no volvió a aparecer después de la suspensión del tratamiento con prednisona. La sinovitis, el
dolor muscular y la inflamación fueron más lentos en su resolución, y se consideró
la posibilidad de un síndrome doloroso regional crónico. Afortunadamente, estos
síntomas también se resolvieron en unas
semanas, y el paciente pudo regresar a la universidad a tiempo para el
semestre de otoño. Síntomas de vasculitis por IgA no han vuelto a aparecer en este paciente.
DIAGNÓSTICO
ANATÓMICO
VASCULITIS
POR IGA.
Fuente:
Traducción de The New England Journal of Medicine
Case 14-2017: A
20-Year-Old Man with Pain and Swelling of the Left Calf and a Purpuric Rash
Eli M. Miloslavsky, M.D., John H. Stone, M.D., M.P.H.,
and Andrea P. Moy, M.D.
References
1. Carlson JA, Chen KR. Cutaneous pseudovasculitis.
Am J Dermatopathol 2007; 29:
44-55.
2. Hoffman GS, Kerr GS, Leavitt RY, et al.
Wegener granulomatosis: an analysis of
158 patients. Ann Intern Med 1992; 116:
488-98.
3. Finkielman JD, Lee AS, Hummel AM,
et al. ANCA are detectable in nearly all
patients with active severe Wegener’s
granulomatosis. Am J Med 2007; 120(7):
643.e9-14.
4.
Jennette JC, Falk RJ, Bacon PA, et al.
2012 Revised International Chapel Hill
Consensus Conference nomenclature of
vasculitides. Arthritis Rheum 2013; 65: 1-11.
5. Xu LY, Esparza EM, Anadkat MJ,
Crone KG, Brasington RD. Cutaneous manifestations
of vasculitis. Semin Arthritis
Rheum 2009; 38: 348-60.
6. Chen KR, Carlson JA. Clinical approach
to cutaneous vasculitis. Am J Clin
Dermatol
2008; 9: 71-92.
7.
Curtis JR, Patkar N, Xie A, et al. Risk
of serious bacterial infections among
rheumatoid arthritis patients exposed to
tumor necrosis factor alpha antagonists.
Arthritis Rheum 2007; 56: 1125-33.
8. Pagnoux C, Cohen P, Guillevin L. Vasculitides
secondary to infections. Clin Exp
Rheumatol 2006; 24: Suppl 41: S71-S81.
9. Poon SH, Baliog CR Jr, Sams RN,
Robinson-Bostom L, Telang GH, Reginato
AM. Syndrome of cocaine-levamisoleinduced
cutaneous vasculitis and immunemediated
leukopenia. Semin Arthritis
Rheum 2011; 41: 434-44.
10. Sy A, Khalidi N, Dehghan N, et al.
Vasculitis in patients with inflammatory
bowel diseases: a study of 32 patients and
systematic review of the literature. Semin
Arthritis Rheum 2016; 45: 475-82.
11. Ramos-Casals M, Stone JH, Cid MC,
Bosch X. The cryoglobulinaemias. Lancet
2012; 379: 348-60.
12. Calvo-Río V, Loricera J, Mata C, et al.
Henoch-Schönlein purpura in northern
Spain: clinical spectrum of the disease in
417 patients from a single center. Medicine
(Baltimore)
2014; 93: 106-13.
13.
Hočevar A, Rotar Z, Ostrovršnik J, et
al. Incidence of IgA vasculitis in the adult
Slovenian population. Br J Dermatol 2014;
171: 524-7.
14. Mahr AD, Neogi T, Merkel PA. Epidemiology
of Wegener’s granulomatosis:
lessons from descriptive studies and analyses
of genetic and environmental risk determinants.
Clin Exp Rheumatol 2006; 24:
Suppl 41: S82-S91.
15.
Blanco R, Martínez-Taboada VM,
Rodríguez-Valverde
V, García-Fuentes M,
González-Gay
MA. Henoch-Schönlein purpura
in adulthood and childhood: two
different expressions of the same syndrome.
Arthritis Rheum 1997; 40: 859-64.
16. Trapani S, Micheli A, Grisolia F, et al.
Henoch Schonlein purpura in childhood:
epidemiological and clinical analysis of
150 cases over a 5-year period and review
of literature. Semin Arthritis Rheum 2005;
35: 143-53.
17. Shet A, Kaplan EL. Clinical use and
interpretation of group A streptococcal
antibody tests: a practical approach for
the pediatrician or primary care physician.
Pediatr Infect Dis J 2002; 21: 420-30.
18. Weiss PF, Klink AJ, Luan X, Feudtner
C. Temporal association of Streptococcus,
Staphylococcus, and parainfluenza pediatric
hospitalizations and hospitalized cases
of Henoch-Schönlein purpura. J Rheumatol
2010;
37: 2587-94.
19.
Marques I, Lagos A, Reis J, Pinto A,
Neves B. Reversible Henoch-Schönlein purpura
complicating adalimumab therapy.
J Crohns Colitis 2012; 6: 796-9.
20. Marzano AV, Borghi A, Stadnicki A,
Crosti C, Cugno M. Cutaneous manifestations
in patients with inflammatory bowel
diseases: pathophysiology, clinical features,
and therapy. Inflamm
Bowel Dis
2014;
20: 213-27.
21. Eloy
P, Leruth E, Goffart Y, et al. Sinonasal
involvement as a rare extraintestinal
manifestation of Crohn’s disease. Eur
Arch Otorhinolaryngol 2007; 264: 103-8.
22. Hung T-Y, Liu MC, Hsu CF, Lin YC.
Facial edema as the initial presentation of
Henoch-Schonlein purpura in a 5-year-old
boy. Pediatr Emerg Care 2009; 25: 31-2.
23. Paydary K, Emamzadeh Fard S, Mahboubi
AH, Ziaee V, Moradinejad MH, Kajbafzadeh
AM. Penile skin involvement as
the first presentation of Henoch-Schon- lein purpura
report of nine cases and review
of literature. Iran J Pediatr 2015;
25(4):
e2177.
24.
Miyazawa T, Sugimoto K, Fujita S, et al.
A patient with Henoch-Schönlein purpura
manifesting unusual symptoms and clinical
course. J Clin Rheumatol 2010; 16: 338-
40.
25. Carlson JA, Chen KR. Cutaneous vasculitis
update: small vessel neutrophilic
vasculitis syndromes. Am J Dermatopathol
2006; 28: 486-506.
26. Russell JP, Gibson LE. Primary cutaneous
small vessel vasculitis: approach to
diagnosis and treatment. Int J Dermatol
2006; 45: 3-13.
27. Carlson JA, Ng BT, Chen KR. Cutaneous
vasculitis update: diagnostic criteria,
classification, epidemiology, etiology,
pathogenesis,
evaluation and prognosis.
Am J Dermatopathol 2005; 27: 504-28.
28. Gupta S, Handa S, Kanwar AJ, Radotra
BD, Minz RW. Cutaneous vasculitides:
clinico-pathological correlation. Indian J
Dermatol Venereol Leprol 2009; 75: 356-
62.
29. Harrist TJ, Mihm MC. Cutaneous immunopathology:
the diagnostic use of direct
and indirect immunofluorescence
techniques in dermatologic disease. Hum
Pathol 1979; 10: 625-53.
30. Hawryluk EB, Linskey KR, Duncan
LM, Nazarian RM. Broad range of adverse
cutaneous eruptions in patients on TNFalpha
antagonists. J Cutan Pathol 2012;
39: 481-92.
31. Linskey KR, Kroshinsky D, Mihm MC
Jr, Hoang MP. Immunoglobulin-A–associated
small-vessel vasculitis: a 10-year experience
at the Massachusetts General
Hospital. J
Am Acad Dermatol 2012; 66:
813-22.
32.
Ronkainen J, Koskimies O, Ala-Houhala
M, et
al. Early prednisone therapy in
Henoch-Schönlein purpura: a randomized,
double-blind, placebo-controlled trial.
J Pediatr 2006; 149: 241-7.











































