En 1905, Starling acuñó el término hormona para describir la secretina, una sustancia secretada por el intestino delgado al torrente sanguíneo para estimular la secreción pancreática. La endocrinología deriva de endon ("dentro") + krinein ("separado"), un término ideado para describir aquellas glándulas que almacenan sus productos dentro de su estructura y los secretan para efectuar una respuesta en tejidos separados. El campo de la endocrinología se ocupa del estudio de las glándulas endocrinas y sus hormonas secretadas.
La comunicación intercelular se logra mediante el
sistema nervioso (señalización eléctrica y química), o el sistema endocrino
(señalización química, hormonas). La regulación del sistema nervioso es rápida
y de corta duración; la regulación por hormonas tiende a ser más lenta y
sostenida. El sistema endocrino mantiene la homeostasis en todo el cuerpo,
regulando diversos sistemas, incluidos el metabolismo, la respiración, la
circulación, la excreción y la reproducción. Es esta diversidad de efectos lo
que hace que los trastornos endocrinos sean particularmente difíciles de
reconocer, diagnosticar y tratar porque pueden tener manifestaciones proteiformes.
Los trastornos clásicos del sistema endocrino están
causados por hiperfunción o hipofunción glandular que dan lugar a estados de
exceso o deficiencia hormonal (tabla 57.1).


Tabla 57.1. Síndromes fisiopatológicos seleccionados y
sus asociaciones hormonales
La hipofunción puede deberse a destrucción glandular
(p. ej., tumor, infección, hemorragia), o problemas de biosíntesis de hormonas.
La hiperfunción generalmente resulta de un tumor o estimulación autoinmune. En
este capítulo se revisan los trastornos seleccionados, junto con los enfoques
diagnósticos. La resistencia a las hormonas, también juega un papel importante
en las enfermedades, particularmente en la diabetes.
Desde el punto de vista del diagnóstico, si un
endocrinólogo sospecha que una hormona está inapropiadamente baja, intentará
estimularla; si es alta, intentará suprimirla (Tabla 57.2).

Tabla 57.2. Causas, Diagnóstico y Tratamiento de la
Hipofunción e Hiperfunción.
El hecho de que no suba o baje normalmente indica una
falla de la glándula o una hiperfunción no regulada. A menudo es posible medir
el nivel de la hormona corriente arriba para determinar la ubicación (central
[pituitaria], o periférica), del problema. Al pasar por este proceso,
capitalizamos la presencia de bucles de retroalimentación. De manera similar,
los tratamientos para los trastornos endocrinos tienen como objetivo reemplazar
la hormona en estados de deficiencia (p. ej., hipotiroidismo) o interferir con
la producción en estados de exceso (p. ej., prolactinoma).
TUMORES PITUITARIOS
Los tumores hipofisarios representan aproximadamente
el 15% de todas las neoplasias intracraneales primarias. La proliferación de
células dentro de la pituitaria puede causar síndromes de exceso hormonal o
comprimir la pituitaria, lo que resulta en una deficiencia hormonal. Los
tumores hipofisarios pueden expandirse e interferir con las estructuras
anatómicas locales, como el quiasma óptico y los senos cavernosos.
PROLACTINOMA
Los tumores secretores de prolactina representan la
mayoría de los tumores hipofisarios funcionales. Más del 90% son microadenomas
benignos (<1 cm) que no aumentan de tamaño.
CARACTERÍSTICAS CLÍNICAS
La hiperprolactinemia provoca galactorrea en
aproximadamente el 80% de las mujeres afectadas. Los altos niveles de
prolactina interfieren con la secreción de la hormona liberadora de
gonadotropina del hipotálamo, lo que provoca amenorrea/oligomenorrea e
infertilidad en las mujeres e hipogonadismo en los hombres. Tanto hombres como
mujeres pueden desarrollar osteoporosis si la hiperprolactinemia no se trata de
forma crónica. Los macroprolactinomas (>1 cm) pueden agrandarse en un tercio
o más durante el embarazo.
EVALUACIÓN
La dopamina inhibe tónicamente la liberación de
prolactina, por lo que los medicamentos que interfieren con la dopamina a
menudo se asocian con hiperprolactinemia. Las fenotiazinas, las butirofenonas,
la metoclopramida, la risperidona, los inhibidores de la monoaminooxidasa, los
antidepresivos tricíclicos, el verapamilo y los inhibidores de la recaptación
de serotonina pueden aumentar los niveles de prolactina. Los tumores hipofisarios
grandes pueden comprimir el tallo hipofisario y alterar la señalización normal
de dopamina, lo que produce hiperprolactinemia. La insuficiencia renal crónica
y el hipotiroidismo primario grave también se asocian con hiperprolactinemia.
Se recomienda la resonancia magnética de la hipófisis cuando un paciente tiene
niveles elevados de prolactina y se han excluido otras causas de
hiperprolactinemia.
TRATAMIENTO
Las indicaciones para el tratamiento incluyeron
galactorrea sintomática, hipogonadismo o infertilidad y macroprolactinoma.
Tanto la cabergolina (que normalmente se administra
dos veces por semana) como la bromocriptina (que se administra dos veces al
día) son eficaces para disminuir los niveles de prolactina y reducir el tamaño
del tumor en >80% de los pacientes. El objetivo del tratamiento es el alivio
sintomático en pacientes con galactorrea, la restauración del eugonadismo o la
fertilidad y la reducción del tamaño del tumor, en el caso de macroprolactinoma.
Tanto los microprolactinomas como los macroprolactinomas
pueden tratarse médicamente; rara vez se requiere cirugía. El tratamiento se
continúa durante al menos 2 años en pacientes con microadenomas y puede
administrarse indefinidamente en pacientes con macroadenomas. Se prefiere la
bromocriptina a la cabergolina en mujeres que están pensando en quedar embarazadas. Los macroadenomas que no se reducen con el agonista de la dopamina
pueden reducirse quirúrgicamente antes de intentar el embarazo. Algunas mujeres
con macroprolactinomas pueden necesitar bromocriptina durante el embarazo.
ACROMEGALIA
La acromegalia es rara y se desarrolla cuando los
somatotropos proliferan y secretan en exceso la hormona del crecimiento. Estos
tumores tienden a crecer de manera lenta e insidiosa y rara vez se asocian con
polisecreción plurihormonal (como la coproducción de prolactina y hormona del
crecimiento). La mayoría de estos tumores son >1 cm (macroadenomas), en el
momento de la presentación.
CARACTERÍSTICAS CLÍNICAS
Las características de la acromegalia son diversas y
generalmente están relacionadas con el crecimiento somático (agrandamiento
acral, maloclusión dentaria, síndrome del túnel carpiano), agrandamiento de tejidos
(macroglosia, hipertrofia prostática, hipertrofia ventricular izquierda, apnea
del sueño, bocio) e interferencia metabólica (diabetes mellitus,
hipertrigliceridemia, hipogonadismo).
EVALUACIÓN
El diagnóstico bioquímico de la acromegalia se realiza
mediante la confirmación de la secreción autónoma de la hormona del crecimiento
durante una prueba de tolerancia a la glucosa oral de 75 g de 2 horas. Los
niveles nadir de la hormona del crecimiento superiores a 1 μg/L son
consistentes con el diagnóstico, especialmente si el paciente también tiene un
nivel elevado del factor de crecimiento similar a la insulina-1 (IGF-1).
TRATAMIENTO
La resección neuroquirúrgica es el tratamiento de
elección para la mayoría de los tumores hipofisarios resecables asociados con
la acromegalia. Las complicaciones como el hipopituitarismo y el riesgo de
recurrencia se correlacionan con el tamaño del tumor. Los tratamientos médicos
para la acromegalia incluyen ligandos del receptor de somatostatina
(octreotida, lanreotida y pasireotida), que suprimen la liberación pituitaria
de la hormona del crecimiento; antagonistas de la dopamina (la cabergolina es
mínimamente efectiva); y el uso de un antagonista del receptor de la hormona
del crecimiento (pegvisomant). La radioterapia se usa para tumores que no
responden, pero se asocia con un alto riesgo de panhipopituitarismo. Los niveles
de IGF-1 se utilizan para controlar la eficacia del tratamiento.
OTROS TRASTORNOS DE LA PITUITARIA
DIABETES INSÍPIDA
La diabetes insípida central es una condición
heterogénea caracterizada por poliuria y polidipsia como resultado de una
deficiencia de arginina vasopresina. En muchos pacientes se debe a la
destrucción o degeneración de las neuronas que se originan en los núcleos
supraóptico y paraventricular del hipotálamo. Las tres causas principales de
diabetes insípida (trauma/cirugía, tumores e idiopática), representan fracciones
de casos aproximadamente iguales.
CARACTERÍSTICAS CLÍNICAS
La arginina vasopresina disminuida o ausente causa
poliuria o polidipsia al disminuir la capacidad del paciente para concentrar la
orina. Estos pacientes generalmente tienen nocturia severa y consumen de 3 a 20
L de líquido al día. La diabetes insípida nefrogénica se caracteriza por una
disminución en la capacidad de concentrar la orina debido a una resistencia a
la acción de la arginina vasopresina en el riñón.
EVALUACIÓN
La diabetes insípida se diagnostica cuando la orina se
diluye de manera inapropiada (gravedad específica de la orina de <1,005 y
una osmolalidad de la orina <200 mOsm/kg), en un paciente con una osmolalidad
sérica alta (generalmente >290 mOsm/kg). Debido a que muchos pacientes
pueden mantener una osmolaridad normal con acceso al agua, a menudo es
necesaria una prueba de privación de agua, para documentar la anomalía y excluir
la polidipsia primaria. Se puede administrar un análogo de vasopresina a los pacientes
al final de una prueba de privación de agua para descartar diabetes insípida
nefrogénica. Los pacientes con diabetes insípida central deben someterse a
imágenes hipofisarias con una resonancia magnética.
TRATAMIENTO
La diabetes insípida se trata con análogos de
vasopresina, administrados por vía subcutánea, nasal u oral, junto con fácil
acceso al agua.
HIPOFISITIS LINFOCITICA
La hipofisitis linfocítica es una enfermedad
autoinmune poco común en la que la glándula pituitaria está infiltrada por linfocitos,
células plasmáticas y macrófagos, lo que generalmente causa una función
alterada. La hipofisitis linfocítica se asocia principalmente con la última
etapa del embarazo o el puerperio, aunque puede ocurrir en mujeres no
embarazadas (algunas de las cuales pueden ser posmenopáusicas), y en hombres.
Este trastorno puede tener un origen autoinmune y está asociado con otros
trastornos autoinmunes, especialmente la tiroiditis autoinmune. Las pruebas de
anticuerpos antinucleares y factor reumatoide a menudo son positivas y la tasa
de sedimentación de eritrocitos puede estar elevada. La cefalea, el deterioro
del campo visual y, más raramente, la diplopía son causados por agrandamiento
hipofisario extraselar, con compresión del quiasma óptico y/o invasión de los
senos cavernosos. La deficiencia de la hormona adrenocorticotrópica (ACTH) (que
produce hipoadrenalismo secundario) es la manifestación endocrina más temprana
y más frecuente. La resonancia magnética suele demostrar agrandamiento
hipofisario simétrico extraselar, y pérdida del punto brillante hipofisario
posterior. El tratamiento es sintomático, con efectos compresivos reducidos
mediante el tratamiento con corticosteroides (20 a 60 mg/día de prednisona) y/o
descompresión quirúrgica. El pronóstico para la recuperación hormonal es malo.
HIPOTIROIDISMO
PRESENTACIÓN DEL HIPOTIROIDISMO
El hipotiroidismo afecta aproximadamente al 2 % de las
mujeres adultas, y aproximadamente al 0,2 % de los hombres adultos; el
hipotiroidismo subclínico es sustancialmente más común. El hipotiroidismo
primario se refiere a la insuficiencia tiroidea intrínseca y representa el 99%
de los casos. El hipotiroidismo secundario se refiere al hipotiroidismo
resultante de una disfunción pituitaria. La causa más común de hipotiroidismo primario
en áreas con suficiente yodo, es la tiroiditis autoinmune crónica (Hashimoto).
Es más común entre las mujeres mayores y generalmente es permanente. La
tiroidectomía, el tratamiento con yodo radiactivo y la radioterapia externa son
otras causas frecuentes de hipotiroidismo. Tanto la deficiencia como el exceso
de yodo pueden causar hipotiroidismo. La deficiencia de yodo es un tipo de
hipotiroidismo asociado con bocio. Es la causa más común de hipotiroidismo
en todo el mundo, pero es bastante poco común en los Estados Unidos, donde se
agrega yodo a la sal. La administración aguda de yodo suprime la síntesis de
tiroxina (efecto Wolf-Chaikof); sin embargo, los pacientes recuperan su función
tiroidea después de unos pocos días de tratamiento. Otros medicamentos que
causan hipotiroidismo incluyen medicamentos antitiroideos (p. ej., metimazol,
propiltiouracilo), amiodarona, litio e interferón-α.
El hipotiroidismo puede ser transitorio cuando se
relaciona con tiroiditis. La inflamación de la glándula tiroides puede ser
dolorosa o completamente indolora. El hipotiroidismo que ocurre después del
parto es una de las presentaciones más comunes de tiroiditis. Aunque el
hipotiroidismo transitorio se resuelve en una mayoría significativa dentro de
los 3 meses, puede durar hasta 6 meses.
CARACTERÍSTICAS CLÍNICAS
Los pacientes con hipotiroidismo pueden presentar una
variedad de síntomas inespecíficos. Algunas de las características más comunes
incluyen piel seca, intolerancia al frío, aumento de peso, estreñimiento, menorragia
y fatiga. El cuadro clínico del hipotiroidismo ahora es mucho más leve desde
que la detección se hizo más común. Los signos más comunes en pacientes con
hipotiroidismo moderado a severo incluyen bradicardia, retraso en la fase de
relajación de los reflejos tendinosos profundos, hinchazón periorbitaria y
vello áspero. El mixedema se refiere a la apariencia de la piel y los tejidos
subcutáneos en un paciente con hipotiroidismo grave.
EVALUACIÓN
Los pacientes con insuficiencia tiroidea primaria
tendrán un nivel elevado de hormona estimulante de la tiroides (TSH) y una
concentración baja de tiroxina. Se debe sospechar hipotiroidismo central si la
TSH es normal o baja y el nivel de tiroxina es bajo.
TRATAMIENTO
Los pacientes con un nivel elevado de TSH deben ser
tratados con tiroxina de reemplazo, con un objetivo de TSH de entre 1 y 2. La
levotiroxina tiene una vida media larga y el tratamiento una vez al día da como
resultado un nivel sérico de tiroxina casi constante. Como resultado de las
variaciones en el contenido de tiroxina de las formulaciones individuales, está
indicada la reevaluación de la adecuación del reemplazo si se cambia la
formulación. La dosis media de reemplazo de tiroxina es de 1.6 μg/kg
(generalmente 75 a 112 μg/día en mujeres y 125 a 200 μg/día en hombres). Se
debe iniciar una dosis más baja (como 50 μg diarios) en los ancianos, y
aumentarla según sea necesario. Los pacientes obesos requieren dosis que son
aproximadamente un 20% más altas. Los fármacos que interfieren con la absorción
de levotiroxina incluyen colestiramina, carbonato de calcio y sulfato ferroso.
Los pacientes que reciben reemplazo de estrógeno también requieren una dosis
más alta de levotiroxina debido al aumento de los niveles de globulina
transportadora de tiroxina.
Algunos pacientes solicitan el reemplazo combinado de
liotironina y levotiroxina, pero no está respaldado por el balance de los datos
de los ensayos clínicos. Si se instituye, 25 μg de levotiroxina pueden
reemplazarse con 5 μg de liotironina. No se recomienda la tiroides desecada, la
liotironina sola ni otras preparaciones para la tiroides.
El hipotiroidismo transitorio puede ser difícil de
distinguir de la tiroiditis de Hashimoto. Es razonable intentar reducir la
dosis de levotiroxina en un 50% después de aproximadamente 3 meses en pacientes
con sospecha de hipotiroidismo transitorio. Si la TSH medida 6 semanas después
aumenta, entonces se restablece la dosis inicial; si la TSH es estable,
entonces se puede retirar la tiroxina y volver a controlar la TSH.
Los pacientes con una TSH elevada y un nivel de
tiroxina normal tienen más probabilidades de tener hipotiroidismo subclínico.
Las pruebas para detectar la presencia de anticuerpos contra la peroxidasa
tiroidea pueden ser útiles en estos pacientes porque predicen la progresión
hacia el hipotiroidismo permanente. Estos pacientes deben ser monitoreados por
el desarrollo de hipotiroidismo más severo, o se puede iniciar la levotiroxina
desde el principio.
Los pacientes con hipotiroidismo central deben tener
su dosis de tiroxina ajustada al nivel de tiroxina libre.
HIPOTIROIDISMO DURANTE EL EMBARAZO
Los requerimientos de levotiroxina aumentan ya en la
quinta semana de gestación. Dada la importancia del eutiroidismo materno para
el desarrollo cognitivo fetal normal, las mujeres con hipotiroidismo deben
aumentar su dosis de levotiroxina en aproximadamente un 30% tan pronto como se
confirme el embarazo. Posteriormente, se deben controlar los niveles séricos de
tirotropina y ajustar la dosis de levotiroxina en consecuencia. El objetivo del
reemplazo de tiroxina es mantener la TSH en el rango de referencia específico
del trimestre (por debajo de 2,5 mU/L en el primer trimestre y por debajo de 3
mU/L en el segundo y tercer trimestre) utilizando mediciones realizadas cada 4
a 6 semanas.
HIPERTIROIDISMO
El hipertiroidismo puede ser causado por una mayor
producción de hormona tiroidea (como en la enfermedad de Graves, o un nódulo
autónomo), o una mayor liberación de hormona tiroidea preformada (como en la
tiroiditis). El hipertiroidismo también puede deberse a un reemplazo excesivo
con hormona tiroidea exógena, hipertiroidismo ectópico o estimulación no
regulada del receptor de TSH (como en la enfermedad trofoblástica o un adenoma
hipofisario secretor de TSH).
CARACTERÍSTICAS CLÍNICAS
La presentación del hipertiroidismo puede ser muy
variable, especialmente en personas mayores. Los síntomas característicos
incluyen ansiedad, temblor, palpitaciones, intolerancia al calor, insomnio,
oligomenorrea y pérdida de peso, a pesar de un aumento del apetito. Los signos
típicos incluyen taquicardia, hipertensión sistólica, temblor, retracción
palpebral, retraso palpebral, piel caliente e hiperreflexia. La presencia de
bocio dependerá de la causa del hipertiroidismo. Un solo nódulo palpable o
múltiples nódulos pueden indicar un adenoma tiroideo autónomo o un bocio
multinodular como origen, respectivamente; una glándula tiroides dolorosa y
sensible sugiere tiroiditis granulomatosa. Los signos que sugieren enfermedad
de Graves incluyen bocio, soplo tiroideo, exoftalmos, edema periorbitario y
mixedema pretibial.
EVALUACIÓN
El diagnóstico de hipertiroidismo se confirma mediante
pruebas bioquímicas de los niveles de tiroxina y TSH. Un nivel elevado de
tiroxina con TSH suprimida caracteriza el hipertiroidismo manifiesto. Los
pacientes con hipertiroidismo subclínico pueden tener un nivel de tiroxina
normal y TSH suprimida. Los pacientes ocasionales muestran toxicosis por
triyodotironina con un nivel normal de tiroxina y un nivel elevado de triyodotironina.
Los títulos de autoanticuerpos activadores dirigidos contra el receptor de
tirotropina están presentes en la enfermedad de Graves. El hipertiroidismo
inducido por TSH y la resistencia a la hormona tiroidea, cada uno caracterizado
por niveles elevados de tiroxina total y libre, y un nivel inapropiadamente
elevado de TSH, son muy raros. Un nivel elevado de tiroxina total con un nivel
normal de tiroxina libre y TSH, generalmente se atribuye a anomalías en las
proteínas de unión a la tiroides en pacientes clínicamente eutiroideos.
Cuando la etiología del hipertiroidismo no está clara,
se puede realizar un estudio de captación de yodo radiactivo en la tiroides. La
enfermedad de Graves se caracteriza por una captación alta, mientras que la
tiroiditis se asocia con una captación baja.
TRATAMIENTO
El metimazol es el fármaco antitiroideo preferido en
los Estados Unidos. El uso de propiltiouracilo está limitado a mujeres
embarazadas. La glándula tiroides concentra activamente estos agentes y su
efecto principal es inhibir la síntesis de hormona tiroidea al interferir con
la yodación de los residuos de tirosina en la tiroglobulina mediada por la
peroxidasa tiroidea, un paso crítico en la síntesis de tiroxina y
triyodotironina. El propiltiouracilo, también bloquea la conversión de tiroxina
en triyodotironina dentro de la tiroides y los tejidos periféricos, aunque la
importancia clínica de esta función es incierta. No es necesario ajustar la
dosis en pacientes con insuficiencia renal o hepática, niños o ancianos.
La dosis inicial habitual de metimazol es de 20 mg/día
como dosis única diaria, y la dosis inicial habitual de propiltiouracilo es de
100 mg administrados tres veces al día. Después de la dosificación inicial, se
sugiere realizar pruebas de seguimiento de la función tiroidea aproximadamente
cada 6 semanas hasta que las pruebas de función tiroidea se normalicen. Muchos
pacientes pueden finalmente controlarse con una dosis baja. La frecuencia de
las pruebas se puede reducir a cada 6 meses con el tiempo. Luego de una
discusión sobre el riesgo de recaída, los medicamentos antitiroideos se pueden
retirar después de 12 a 18 meses para determinar si se requiere un tratamiento
continuo.
Las reacciones cutáneas a los medicamentos
antitiroideos son bastante comunes y generalmente leves. El medicamento debe
suspenderse en pacientes que se quejan de artralgias porque esto puede ser una
presentación de una poliartritis migratoria transitoria asociada con el uso de
medicamentos antitiroideos. Los efectos secundarios más temidos de los fármacos
antitiroideos son la agranulocitosis (especialmente con metimazol, visto en 1
de cada 270 pacientes tratados) y la insuficiencia hepática (especialmente con
propiltiouracilo). El fármaco debe suspenderse si el recuento de granulocitos es
<1000/mm3.
Los tratamientos actuales para la enfermedad de Graves
incluyen fármacos antitiroideos, yodo radioactivo y cirugía. El tratamiento
inicial suele incluir un fármaco antitiroideo como metimazol 20 mg una vez al
día. Los betabloqueantes pueden proporcionar alivio sintomático. Para pacientes
con hipertiroidismo más severo, el tratamiento con yodo puede proporcionar un
alivio rápido de los síntomas al prevenir la liberación de tiroxina de la
tiroides. Un enfoque típico sería prescribir tres gotas de solución saturada de
yoduro de potasio tres veces al día durante un máximo de 10 días. El yodo
radiactivo se puede administrar como tratamiento primario para pacientes con
hipertiroidismo de Graves, aunque muchos médicos esperarán hasta la primera
recaída antes de ofrecer este enfoque. El hipertiroidismo puede exacerbarse por
un corto tiempo con el tratamiento con yodo radiactivo. En pacientes con
enfermedad cardiaca o en ancianos en quienes tal exacerbación sería riesgosa,
puede ser útil el pretratamiento con fármacos antitiroideos. La cirugía
generalmente se reserva para pacientes con bocio obstructivo.
El hipertiroidismo asociado con adenomas tiroideos
tóxicos se puede tratar con medicamentos antitiroideos y un bloqueador beta.
Estos nódulos autónomos no se resuelven espontáneamente; por tanto, suele estar
indicado un tratamiento más definitivo una vez controlados los síntomas
iniciales. Para la mayoría de los pacientes, se prefiere el yodo radiactivo a
la cirugía (porque tiende a atacar solo el tejido hiperactivo, lo que resulta
en un bajo riesgo a largo plazo de hipotiroidismo), pero es menos probable que
sea eficaz en pacientes con nódulos grandes o múltiples.
HIPERTIROIDISMO DURANTE EL EMBARAZO
Las mujeres que desarrollan hipertiroidismo durante el
embarazo tienen un mayor riesgo de aborto espontáneo, parto prematuro, muerte
fetal y preeclampsia. Los cambios en la globulina transportadora de hormonas
tiroideas (generalmente se duplica), los niveles de gonadotropina coriónica
humana (que pueden imitar las acciones de la TSH) y la fisiología endógena
(respuesta alterada de la TSH) pueden complicar la evaluación bioquímica de la
función tiroidea en el embarazo. Debido a que el yodo radiactivo está
absolutamente contraindicado durante el embarazo, los medicamentos
antitiroideos son el tratamiento preferido para las mujeres embarazadas con
hipertiroidismo. Las dosis deben reducirse al mínimo para prevenir el
hipotiroidismo fetal porque tanto el propiltiouracilo como el metimazol pueden
atravesar la placenta. El metimazol puede estar asociado con anomalías
congénitas que incluyen aplasia cutis y atresia coanal o esofágica. El
propiltiouracilo se ha asociado con insuficiencia hepática grave. En general,
el tratamiento se administra hasta el punto en que se permite que persista el
hipertiroidismo leve. La función tiroidea baja al nacer se encuentra en
aproximadamente la mitad de los recién nacidos cuyas madres recibieron un
fármaco antitiroideo; la inteligencia última parece ser normal. Tanto el
metimazol como el propiltiouracilo están aprobados para madres lactantes por la
Academia Estadounidense de Pediatría, aunque aparecen en la leche materna en
cantidades mínimas. Debido al potencial de hepatotoxicidad asociada con el
propiltiouracilo, las madres lactantes pueden preferir el metimazol.
NÓDULOS TIROIDEOS
En los Estados Unidos, entre el 1% y el 5% de la
población adulta tiene un nódulo tiroideo palpable, pero entre el 20% y el 70%
tiene nódulos detectables en la ecografía. Aproximadamente del 5% al 15% de
estos son malignos, pero en general, es poco probable que los microcarcinomas de <1 cm sean clínicamente significativos. Otras causas de nódulos incluyen quiste tiroideo, nódulo coloide, área focal de tiroiditis y neoplasias
foliculares benignas. Los nódulos pueden ser solitarios o múltiples. El riesgo
de cáncer no es menor cuando los nódulos son múltiples.
EVALUACIÓN
La historia puede proporcionar información útil para
el pronóstico. El crecimiento rápido o la presencia de antecedentes familiares
de carcinoma de tiroides o neoplasia endocrina múltiple (NEM) aumenta el riesgo
de que un nódulo sea canceroso. El riesgo aumenta de forma más moderado si la
edad es < 30 años o > 60 años, si el paciente es varón, si hay
antecedentes de irradiación de cabeza y cuello, si el nódulo mide > 4 cm o
tiene captación aumentada en la tomografía por emisión de positrones (PET) o si
hay síntomas locales como disfagia, ronquera o tos. Los signos de alto riesgo
incluyen un nódulo duro, la presencia de linfadenopatía regional o ronquera.
Un nivel de TSH suprimido sugiere un nódulo
hiperfuncionante benigno. La exploración con radionucleótidos utiliza yodo 123,
yodo 131 o pertecnetato de tecnecio 99m para detectar si un nódulo funciona o
no. Una exploración no puede determinar con precisión el tamaño de un nódulo
tiroideo.
Los nódulos hiperfuncionantes son tan raramente
malignos que si se suprime la TSH, se puede diferir la aspiración con aguja
fina (AAF).
Un nivel de TSH normal o alto no elimina la necesidad
de una mayor investigación. La ecografía se puede realizar en el consultorio y
aumenta la sensibilidad y especificidad del resultado de la AAF. La ecografía
puede detectar características de alto riesgo como composición sólida,
hipoecogenicidad, microcalcificaciones, márgenes irregulares, forma más alta
que ancha y evidencia de linfadenopatía local.
Se recomienda AAF para nódulos ≥1 cm y cualquiera de
las características ecográficas de alto riesgo. Si ninguna de estas
características está presente, se recomienda AAF para nódulos ≥1,5 cm de
tamaño. La biopsia no es necesaria para los nódulos puramente quísticos. Los
pacientes que se someten a AAF normalmente no necesitan suspender la aspirina o
los anticoagulantes. El procedimiento es seguro y bien tolerado.
Los nódulos quísticos se pueden drenar después de
aspirar cualquier parte sólida, aunque la mayoría recurre; el uso de
esclerosantes como el etanol y la tetraciclina ha sido decepcionante.
TRATAMIENTO
Los nódulos benignos pueden permanecer in situ. Se
sugiere repetir la ecografía después de 12 a 24 meses para evaluar el
crecimiento significativo o el desarrollo de nuevas características ecográficas
sospechosas. Rara vez se recomienda la supresión con levotiroxina porque el
potencial de daño (pérdida ósea y fibrilación auricular) supera el modesto
beneficio de la supresión del crecimiento.
Los nódulos con citología indeterminada o neoplasia
folicular, fríos en la gammagrafía tiroidea pueden ser benignos o malignos. Se
pueden considerar las pruebas moleculares para guiar las opciones quirúrgicas.
Los nódulos malignos generalmente deben extirparse. La extensión de la cirugía
y la necesidad de ablación tiroidea posoperatoria con yodo 131 dependerán de
múltiples factores clínicos. La recurrencia de la enfermedad después de la tiroidectomía
puede detectarse mediante mediciones de tiroglobulina.
Los aspirados no diagnósticos ocurren alrededor del
10% de las veces. Si una segunda muestra nuevamente no es diagnóstica, entonces
es apropiada la derivación para escisión quirúrgica.
CÁNCER DE TIROIDES
Los cánceres de tiroides papilar y folicular
representan aproximadamente el 80 % y el 10 % de todos los cánceres de la
glándula tiroides, respectivamente. Metástasis, linfomas y cánceres medulares y
anaplásicos conforman el resto de los casos. Las supervivencias a 10 años son
muy diferentes para estos tumores, oscilando entre el 98 % para el papilar, el
92 % para el folicular y <10 % para el carcinoma anaplásico.
CARACTERÍSTICAS CLÍNICAS
La mayoría de los tumores se detectan de manera
incidental o se palpa un nódulo. Las características de alto riesgo de un
nódulo incluyen crecimiento rápido, antecedentes familiares de carcinoma de
tiroides o neoplasia endocrina múltiple, edad <30 años o >60 años, sexo
masculino y si hay antecedentes de irradiación de cabeza y cuello. Los síntomas
locales como disfagia, ronquera o tos son preocupantes.
EVALUACIÓN
Los cánceres de tiroides casi siempre se diagnostican
mediante AAF. Los cánceres diferenciados se clasifican según la clasificación
TNM para estimar la mortalidad. En esta clasificación, todos los pacientes
menores de 45 años se encuentran en el estadio 1 a menos que tengan metástasis
a distancia (estadio 2). Para los pacientes mayores, el estadio depende del
tamaño, la presencia y la lateralidad de la afectación de los ganglios
linfáticos, y si se han detectado metástasis a distancia. Se utiliza un sistema
de estratificación diferente que se basa en los hallazgos clinicopatológicos
(histología, marcadores moleculares, grado de invasión, enfermedad residual,
afectación de los ganglios linfáticos) para estimar el riesgo de recurrencia de
la enfermedad.
TRATAMIENTO
La tiroidectomía total se recomienda para los cánceres
tiroideos diferenciados de tamaño ≥4 cm, con extensión extratiroidea o con
metástasis a ganglios linfáticos o sitios distantes. La lobectomía unilateral
se puede considerar para tumores intratiroideos más pequeños. La disección
regional del cuello está indicada si una ecografía preoperatoria del cuello
muestra evidencia de adenopatía. Después de la cirugía, con frecuencia se
requiere terapia con levotiroxina para prevenir el hipotiroidismo sintomático y
reducir la posible estimulación de la tirotropina del crecimiento tumoral. El
nivel objetivo de tirotropina generalmente está por debajo o en la mitad
inferior del rango normal; los objetivos más bajos se utilizan para la
enfermedad avanzada. La ablación con yodo radiactivo se reserva para pacientes
de alto riesgo y algunos de riesgo intermedio después de una tiroidectomía
total. La respuesta al tratamiento se determina principalmente mediante
ecografía del cuello y mediciones de tiroglobulina sérica para identificar la
enfermedad residual. La radioterapia de haz externo adyuvante y algo de
quimioterapia se usan ocasionalmente para casos refractarios.
El cáncer de tiroides anaplásico es casi siempre
fatal. El cáncer generalmente se trata quirúrgicamente. Si no son resecables,
algunos de estos tumores responden a la doxorrubicina y la radioterapia de haz
externo para el control local.
OSTEOPOROSIS
La osteoporosis es un problema común que afecta en
última instancia a la mitad de todas las mujeres posmenopáusicas y
aproximadamente a una cuarta parte de los hombres. Se caracteriza por una masa
ósea baja y un mayor riesgo de fractura. Las fracturas ocurren debido al
deterioro cualitativo y cuantitativo del esqueleto trabecular y cortical. La
calidad ósea no se puede medir clínicamente, pero la densidad mineral ósea se
puede medir fácilmente mediante densitometría ósea.
CARACTERÍSTICAS CLÍNICAS
La osteoporosis es asintomática hasta que se produce
la fractura. La fractura vertebral es la más común, y la mayoría son
asintomáticas. Las fracturas de cadera y las fracturas radiales son comunes.
EVALUACIÓN
La mayoría de las fracturas de cadera, muñeca y cuerpo
vertebral sin traumatismo son indicativas de osteoporosis. En la densitometría,
la osteopenia se define como una masa ósea que está entre 1 y 2,5 desviaciones
estándar por debajo de la masa ósea máxima media de la población de control,
también conocida como puntuación T. La osteoporosis se define como un valor de
masa ósea > 2,5 desviaciones estándar por debajo de la masa ósea máxima de
una población de control. En general, se toman imágenes de la cadera y la
columna. En mujeres blancas posmenopáusicas, el riesgo relativo de fractura
aumenta en un factor de 1,5 a 3 por cada disminución de 1 en la puntuación T.
La densitometría también proporciona la desviación de la masa ósea de los
controles de la misma edad y la notifica como una puntuación Z. La puntuación Z
determina si alguna pérdida ósea es sustancialmente mayor de lo esperado para
la edad. Una evaluación básica con baja densidad ósea comprende un perfil
bioquímico, enzimas hepáticas, fosfatasa alcalina, 25-hidroxivitamina D y un
hemograma completo. Los pacientes con puntajes Z bajos deben someterse a una
evaluación de causas secundarias de pérdida ósea, como hiperparatiroidismo,
hipertiroidismo, mieloma y malabsorción. Los marcadores de recambio óseo (como
el telopéptido N-terminal urinario o el telopéptido C-terminal sérico) pueden
ser útiles para determinar la necesidad de tratamiento en casos dudosos.
TRATAMIENTO
Una estrategia inicial óptima para pacientes con baja
masa ósea incluye suplementos de calcio (1000 a 1200 mg elementales) y vitamina
D (400 a 800 UI diarias) cuando sea apropiado, actividad física con carga de
peso (30 minutos al menos tres veces por semana) y asesoramiento para dejar de
fumar si es necesario. Los pacientes con cualquiera de los siguientes cuatro
criterios califican para la intervención farmacológica: (1) antecedentes de
fractura de cadera o vertebral; (2) osteoporosis por densitometría; (3)
cualquier fractura por fragilidad y osteopenia por densitometría; (4)
osteopenia por densitometría con un riesgo a 10 años de una fractura
osteoporótica mayor de más del 20 % o de fractura de cadera de más del 3 %
(calculadora de riesgo en http://www.shef.ac.uk/FRAX/) .
Los bisfosfonatos son el tratamiento de primera línea
recomenddo para pacientes con osteoporosis. El alendronato (70 mg por vía oral
una vez a la semana) y el risedronato (35 mg una vez a la semana o 150 mg una
vez al mes) se administran por vía oral. Zoledronato 5 mg se administra por vía
intravenosa una vez al año. Se ha demostrado que todos reducen el riesgo de
fracturas de cadera y columna. El ibandronato (150 mg una vez al mes por vía
oral o 3 mg por vía intravenosa cada 3 meses) tiene datos más limitados con
respecto a su eficacia.
Los efectos adversos gastrointestinales de los
bisfosfonatos orales son raros si se siguen las instrucciones de
administración. Los bisfosfonatos intravenosos provocan síntomas similares a
los de la gripe a corto plazo. La hipocalcemia puede complicar el tratamiento
con bisfosfonatos si los niveles de vitamina D no son suficientes. La osteonecrosis
de la mandíbula es una complicación rara del uso de bisfosfonatos, que ocurre
una vez cada 10 000 a 100 000 años-paciente. Aunque la mayoría de los casos han
sido en pacientes con cáncer o en pacientes con cánceres tratados con
bisfosfonatos intravenosos, se han observado casos raros en pacientes con
osteoporosis posmenopáusica que toman bisfosfonatos orales.
La hormona paratiroidea sintética (PTH: teriparatida)
está disponible para uso subcutáneo diario y aumenta la densidad mineral ósea y
reduce el riesgo de fracturas. Generalmente se reserva para pacientes con
osteoporosis severa; el tratamiento está limitado a 24 meses. El denosumab es
un medicamento inyectable que se usa para tratar a los pacientes que no toleran
los bisfosfonatos o que no pueden recibir tratamiento con ellos, o que tienen
osteoporosis grave o disfunción renal. En general, la densitometría de control
debe realizarse con una frecuencia no superior a cada 2 años.
HIPERCALCEMIA
CARACTERÍSTICAS CLÍNICAS
Los pacientes con hipercalcemia leve (10.5 a 12 mg/100
ml) suelen estar asintomáticos. Los pacientes con hipercalcemia más grave
pueden presentar síntomas inespecíficos que incluyen náuseas, anorexia,
estreñimiento, dolor abdominal, dolor óseo, fatiga, polidipsia y confusión. Los
niveles de calcio superiores a 14 mg/dL son peligrosos y pueden ser letales.
Los signos de hipercalcemia incluyen arritmias, hipertensión y un intervalo QT
acortado en un electrocardiograma.
EVALUACIÓN
Las dos causas más frecuentes de hipercalcemia son el
hiperparatiroidismo primario y la enfermedad neoplásica, que representan más
del 90% de los casos, y se pueden discriminar en función de la concentración
sérica de PTH. El hiperparatiroidismo primario tiene un curso relativamente
benigno. La osteoporosis y la insuficiencia renal son dos consecuencias
importantes a largo plazo que impulsan una intervención temprana. Se recomienda
la paratiroidectomía en pacientes con PTH elevada e hipercalcemia menores de 50
años o si el calcio es >11,5 mg/dl. Las indicaciones adicionales para la
cirugía incluyen la presencia de cálculos renales, evidencia de insuficiencia
renal, fracturas vertebrales en estudios de imágenes, una puntuación T en la
densitometría ósea de <2.5 e hipercalciuria con análisis bioquímicos de orina
que sugieren un alto riesgo de cálculos renales. Una gammagrafía paratiroidea
preoperatoria con sestamibi o una ecografía pueden ayudar al cirujano endocrino
y limitar la extensión de la cirugía. Los niveles de PTH intraoperatoria
permiten al cirujano estar seguro del éxito del procedimiento antes del cierre
quirúrgico. El calcimimético, cinacalcet, puede usarse para disminuir los
niveles de calcio en pacientes hipercalcémicos que no pueden someterse a
cirugía. Los bisfosfonatos son una opción para los pacientes que no pueden
someterse a cirugía pero tienen evidencia de pérdida ósea.
La insuficiencia renal crónica generalmente aumenta
los niveles de fosfato y disminuye la activación de la vitamina D. Si no se
trata, los niveles elevados prolongados de fosfato y bajos de vitamina D pueden
provocar un aumento de la secreción de PTH y la subsiguiente hipercalcemia.
Esto se denomina hiperparatiroidismo terciario y puede tratarse quirúrgica o
médicamente.
La hipercalcemia de la malignidad suele ser sintomática y
puede ser grave. Los tumores sólidos inducen hipercalcemia al liberar proteína
relacionada con la PTH, que imita la acción de la PTH endógena. La destrucción
ósea por enfermedad metastásica o mieloma también puede inducir hipercalcemia,
a menudo en asociación con un nivel elevado de fosfatasa alcalina.
La vitamina D puede inducir hipercalcemia si se toma
en sobredosis o si hay una acción excesiva de la 1-alfa hidroxilasa que crea la
forma activa de vitamina D. Esta enzima es hiperactiva en pacientes con enfermedad
granulomatosa como la sarcoidosis y responde bien al tratamiento con
glucocorticoides mientras la enfermedad subyacente está siendo tratada. El
consumo de grandes cantidades de calcio o vitamina A rara vez puede provocar
hipercalcemia.
La hipercalcemia hipocalciúrica familiar es una
afección autosómica dominante causada por una mutación en el gen del receptor
sensible al calcio. Los pacientes tienen un curso inocuo caracterizado por
hipercalcemia de leve a moderada, niveles de PTH normales o ligeramente
elevados y excreción urinaria de calcio baja. Estos pacientes no se benefician
de la paratiroidectomía.
TRATAMIENTO
El tratamiento de la hipercalcemia grave incluye la
reposición emergente de líquidos con solución salina y la administración
intravenosa de bisfosfonatos. Inicialmente, la solución salina se administra a
razón de 200 a 300 ml por hora y se ajusta para mantener la producción de orina
entre 100 y 150 ml por hora. Se puede agregar un diurético de asa, pero no
siempre es necesario y puede provocar hipopotasemia e hipomagnesemia. En los
Estados Unidos, el pamidronato y el zoledronato son bisfosfonatos autorizados
para su uso en esta indicación. Se prefiere el zoledronato porque se puede
administrar en menos tiempo (15 minutos en comparación con las 2 horas del
pamidronato) y es más potente.
La hipocalcemia puede ocurrir en pacientes tratados
con bisfosfonatos intravenosos por hipercalcemia de malignidad, aunque la
hipocalcemia sintomática es rara. La calcitonina se caracteriza por una buena
tolerabilidad pero poca eficacia para normalizar el nivel de calcio sérico. Sin
embargo, una ventaja importante de la calcitonina es el inicio agudo del efecto
hipocalcémico (reducción de 1 a 2 mg/dl en 6 h), que contrasta con el efecto
tardío (aproximadamente 2 a 4 días) pero más pronunciado de los bisfosfonatos.
Se administra por vía intramuscular o subcutánea cada 12 horas a dosis de 4
UI/kg (la calcitonina nasal no es eficaz para este fin). Se puede considerar
denosumab en pacientes con insuficiencia renal.
HIPOPARATIROIDISMO
La hipocalcemia resulta de PTH inadecuada, suministro
insuficiente de vitamina D, niveles anormales de magnesio o durante
circunstancias metabólicas como sepsis o pancreatitis. El hipoparatiroidismo se
diagnostica cuando el nivel de PTH es inapropiadamente bajo en un paciente con
hipocalcemia y un nivel de magnesio normal.
CARACTERÍSTICAS CLÍNICAS
Los síntomas neuromusculares como calambres
musculares, entumecimiento y hormigueo peribucal y espasmos musculares son las
características de presentación más típicas de la hipocalcemia. A medida que
los niveles de calcio caen más, pueden ocurrir convulsiones, insuficiencia
cardíaca, broncoespasmo y laringoespasmo. La hipocalcemia crónica puede
provocar cataratas y calcificación de los ganglios basales y se ha asociado con
pseudotumor cerebral.
El hipoparatiroidismo generalmente resulta de una
intervención quirúrgica en el cuello y ocurre hasta en el 5% del total de
cirugías de tiroidectomía. La suficiencia paratiroidea generalmente requiere
una sola glándula paratiroidea remanente. La insuficiencia paratiroidea también
puede ocurrir si las paratiroides acumulan hierro (p. ej., hemocromatosis) o
cobre (enfermedad de Wilson). La destrucción autoinmune de las glándulas
paratiroides generalmente se asocia con el síndrome poliendocrino autoinmune
tipo 1 (que incorpora al menos dos de la tríada de enfermedad de Addison,
hipoparatiroidismo y candidiasis mucocutánea crónica). El hipoparatiroidismo de
aparición temprana que acompaña a la inmunodeficiencia caracteriza el síndrome
de DiGeorge o mutaciones activadoras del gen del receptor sensible al calcio.
El magnesio es esencial para la secreción de PTH, y tanto la hipermagnesemia
como la hipomagnesemia pueden provocar hipocalcemia.
EVALUACIÓN
Se debe calcular el calcio total corregido (calcio
total medido en mg/dL + 0,8 [4 – albúmina sérica en g/dL]) o medir el calcio
ionizado. La evaluación de laboratorio debe incluir medidas de PTH intacta,
25-hidroxivitamina D, 1,25-dihidroxivitamina D, fosfato y magnesio.
TRATAMIENTO
Los pacientes con síntomas graves de hipocalcemia
deben ser tratados con gluconato de calcio intravenoso. El tratamiento a largo
plazo requiere que los pacientes reciban sales de calcio y un metabolito de la
vitamina D, como el calcitriol. Cada uno se titula para obtener niveles
normales. Las tiazidas se pueden usar para reducir la hipercalciuria y prevenir
la nefrolitiasis, particularmente si el nivel de calcio en orina de 24 horas
supera los 250 mg. La reposición de PTH obtuvo recientemente una aprobación
limitada para el tratamiento del hipoparatiroidismo.
FALLO ADRENAL
La enfermedad de Addison se refiere a la insuficiencia
adrenocortical crónica causada por la disfunción de toda la corteza suprarrenal
(incorporando deficiencia de glucocorticoides, mineralocorticoides y esteroides
sexuales). La insuficiencia suprarrenal aguda también puede deberse a cirugía,
hemorragia, invasión tumoral, medicamentos antimicóticos, síndrome de
inmunodeficiencia adquirida o infecciones que afectan ambas glándulas
suprarrenales. La insuficiencia suprarrenal se vuelve sintomática cuando se ha
perdido aproximadamente el 90% de la función suprarrenal.
CARACTERÍSTICAS CLÍNICAS
Los pacientes con crisis suprarrenal aguda presentan
náuseas, vómitos e hipotensión. Puede haber dolor abdominal o en el costado y
fiebre. La insuficiencia suprarrenal crónica se presenta insidiosamente con
debilidad, fatiga, artralgia, anorexia, náuseas y pérdida de peso. El examen
puede revelar hiperpigmentación (como resultado del efecto estimulante de la
adrenocorticotropina sobre los melanocitos) y vitíligo (destrucción autoinmune
de los melanocitos). Otras características del examen incluyen disminución del
vello corporal debido a la pérdida de andrógenos suprarrenales, una característica
que es especialmente evidente en las mujeres.
EVALUACIÓN
Los pacientes con insuficiencia suprarrenal aguda
generalmente estarán hipotensos y pueden tener hiponatremia. La hiperpotasemia
es un indicador de deficiencia de mineralocorticoides. La eosinofilia y la
hipoglucemia pueden acompañar a la crisis suprarrenal.
Una prueba de estimulación con adrenocorticotropina
generalmente comprende la medición de cortisol al inicio del estudio, inyección
intravenosa de 250 μg de adrenocorticotropina, con niveles de cortisol medidos
a los 60 minutos. El nivel de cortisol debe aumentar a más de 18 μg/dl para
establecer una función glucocorticoide normal. Un nivel de cortisol matutino
muy bajo también puede ser indicativo de insuficiencia suprarrenal. La medición
de corticotropina ayuda a diferenciar entre etiologías periféricas y centrales.
Las mediciones simultáneas de aldosterona y renina
pueden determinar la presencia de deficiencia de mineralocorticoides. Deben
medirse los niveles de tirotropina y tiroxina. Los autoanticuerpos
suprarrenales pueden ser útiles para establecer el riesgo de otras enfermedades
autoinmunes.
TRATAMIENTO
El tratamiento agudo comprende de 50 a 100 mg de
hidrocortisona administrados por vía intravenosa. La mejoría clínica debe
seguir dentro de las 6 horas. La reposición de glucocorticoides generalmente
comprende hidrocortisona de 15 a 25 mg diarios en dos o tres dosis divididas,
tituladas según los síntomas. La reposición de mineralocorticoides debe
iniciarse con fludrocortisona para pacientes con insuficiencia suprarrenal
primaria. Las dosis son generalmente de 0,05 a 0,2 mg/d ajustadas a la presión
arterial, el potasio y la actividad de la renina plasmática matutina. Los
pacientes deben recibir glucocorticoides inyectables para uso de emergencia,
como durante vómitos, diarrea y trauma; también deben usar un brazalete o
collar de identificación de emergencia.
NÓDULOS Y TUMORES SUPRARRENALES
Las masas suprarrenales se detectan en aproximadamente
el 5% de todas las exploraciones por imágenes y autopsias abdominales. Al
evaluar las masas suprarrenales, el médico debe considerar si la masa es
maligna y si es hormonalmente activa. La corteza suprarrenal puede producir una
variedad de hormonas y síndromes que incluyen cortisol (síndrome de Cushing),
aldosterona (síndrome de Conn), andrógenos (virilización) y estrógenos
(feminización); la médula suprarrenal genera catecolaminas (feocromocitoma).
EVALUACIÓN
Las características de imagen pueden ayudar a
determinar el riesgo de malignidad. Las características de alto riesgo incluyen
forma irregular, diámetro >4 cm, alto valor de atenuación en la TC (>10
unidades Hounsfield [HU]) y realce no homogéneo con lavado lento después del
contraste intravenoso. La enfermedad metastásica de otra fuente tiende a causar
enfermedad bilateral y tiene una atenuación similar a la del hígado en las
imágenes T1 y una intensidad de señal alta en T2. Los nódulos benignos (que
pueden ser funcionales) tienden a ser redondos, homogéneos y más pequeños
(<4 cm) con baja atenuación en la TC (<10 HU), lavado rápido del
contraste intravenoso e isointensos con el hígado en T1 y T2. Imágenes de
resonancia magnética. Los quistes suprarrenales, el mielolipoma y la hemorragia
suprarrenal suelen distinguirse fácilmente por sus características de imagen
únicas.
CARACTERÍSTICAS CLÍNICAS
El feocromocitoma puede ser sugerido por la presencia
de hipertensión (puede ser crónica o paroxística), antecedentes de
"episodios", de dolor de cabeza, palpitaciones o palidez. Incluso en
ausencia de síntomas, el feocromocitoma siempre debe excluirse antes de
proceder a la cirugía porque los riesgos intraoperatorios de un feocromocitoma
no reconocido son altos. Las metanefrinas séricas son muy sensibles pero no tan
específicas. Dos recolecciones urinarias consecutivas de 24 horas para
metanefrinas totales y catecolaminas brindan evidencia confirmatoria.
El aldosteronismo primario se sugiere por hipertensión
refractaria y, en ocasiones, por hipopotasemia. Una proporción de aldosterona a
renina de >30 es sugestiva. Para obtener un resultado interpretable, los
pacientes no deben estar tomando antagonistas de los receptores de aldosterona
(como espironolactona, eplerenona). Una renina suprimida en un paciente que
toma un inhibidor de la enzima convertidora de angiotensina (ACE) o un
bloqueador del receptor de angiotensina es altamente sugestiva de
hiperaldosteronismo.
Las mediciones de andrógenos y estrógenos no se
realizan de forma rutinaria en ausencia de síntomas o signos sugestivos. La
virilización es sugerida por la calvicie de patrón masculino, la profundización
de la voz y la clitoromegalia en las mujeres. La feminización en los hombres es
sugerida por ginecomastia, disminución de la libido y pérdida de fuerza
muscular.
ALDOSTERONISMO PRIMARIO
El aldosteronismo primario resultante de un adenoma
suprarrenal es una causa reversible de hipertensión; representa al menos el 10%
de las causas de hipertensión resistente. Estos tumores suelen tener un tamaño
<2 cm y son benignos; la mayoría de los adenomas son unilaterales.
CARACTERÍSTICAS CLÍNICAS
La mayoría de los pacientes son asintomáticos o tienen
síntomas mínimos. El dolor de cabeza puede acompañar a la hipertensión severa.
La poliuria, la nicturia y los calambres musculares pueden acompañar a la
hipopotasemia.
EVALUACIÓN
Es común la hipopotasemia espontánea con alcalosis
metabólica y un nivel sérico de sodio en el extremo superior del rango normal o
hipernatremia. La actividad de la renina plasmática está suprimida en casi
todos los pacientes con aldosteronismo primario no tratado y los niveles de
aldosterona plasmática están elevados. Una proporción de aldosterona plasmática
a renina plasmática de >30 con una aldosterona plasmática de >10 ng/dl
por lo general indica aldosteronismo primario. Las concentraciones de
aldosterona no se pueden interpretar en un paciente con espironolactona. Los
diuréticos, los inhibidores de la ECA y los bloqueadores de los receptores de
angiotensina pueden elevar falsamente la actividad de la renina plasmática, lo
que conduce a una relación más baja de aldosterona a actividad de renina
plasmática; por lo tanto, la presencia de actividad de renina plasmática
suprimida en un paciente tratado con un diurético o, especialmente, un
inhibidor de la ACE o un bloqueador del receptor de angiotensina, es un fuerte
predictor de hiperaldosteronismo primario. La prueba más definitiva para
diagnosticar el aldosteronismo primario es una tasa de excreción de aldosterona
urinaria de 24 horas no suprimida durante una carga de sal (de al menos 1
cucharadita de sal al día durante 3 días). Los pacientes mayores de 40 años
generalmente requieren una muestra de la vena suprarrenal para evitar la
resección de un adenoma cortical inocuo.
TRATAMIENTO
Los pacientes con adenoma productor de aldosterona
unilateral confirmado pueden proceder a una suprarrenalectomía unilateral. Los
pacientes con hiperplasia suprarrenal bilateral y los pacientes que rehúsan o
no son candidatos quirúrgicos pueden ser manejados con espironolactona o
eplerenona con un programa de restricción de sal y actividad aeróbica regular.
FEOCROMOCITOMA
Los tumores secretores de catecolaminas pueden dar
lugar a un síndrome clínico dramático y potencialmente mortal. La mayoría de
estos tumores surgen de la médula suprarrenal. Los tumores que surgen fuera de
la glándula suprarrenal se denominan paragangliomas. Estos tumores son raros y
representan menos de 1 de cada 1000 casos de hipertensión.
CARACTERÍSTICAS CLÍNICAS
La tríada clásica incluye cefalea episódica,
palpitaciones y diaforesis. Los episodios pueden ocurrir con una frecuencia tan
baja como una vez al mes o varias veces al día. Ansiedad, náuseas, temblores,
dolor torácico y epigástrico y pérdida de peso son otras características
reportadas. Los pacientes con feocromocitoma suelen ser hipertensos;
aproximadamente la mitad de los pacientes tendrán hipertensión episódica.
EVALUACIÓN
Debido a que pueden ocurrir feocromocitomas y
paragangliomas en asociación con el síndrome de neoplasia endocrina múltiple
tipo 2, en la neurofibromatosis 1, en el síndrome de von Hippel Lindau y en
pacientes con mutaciones de succinato deshidrogenasa, se deben buscar
indicaciones de estos diagnósticos.
Los pacientes con bajo riesgo de feocromocitoma pueden
descartar el diagnóstico mediante la medición de catecolaminas y metanefrinas
en una muestra de orina de 24 horas, una prueba con alta especificidad. Los
pacientes de alto riesgo pueden ser examinados con metanefrinas plasmáticas. La
mayoría de los pacientes con feocromocitomas tienen niveles de metanefrina en
orina o plasma que son al menos tres o cuatro veces superiores al límite
superior normal. Los antidepresivos tricíclicos, levodopa, labetalol, etanol,
sotalol, anfetaminas, buspirona, benzodiazepinas, metildopa y clorpromazina
aumentan las catecolaminas y deben evitarse.
Si el diagnóstico se confirma bioquímicamente, se
recomiendan imágenes de las glándulas suprarrenales con MRI; estas lesiones
suelen tener al menos 3 cm de tamaño y los feocromocitomas son especialmente
hiperintensos en las imágenes potenciadas en T2. La exploración de medicina
nuclear con metayodobencilguanidina 123I o PET se puede realizar si las
imágenes de MR/CT son equívocas.
TRATAMIENTO
Los pacientes con feocromocitoma deben ser tratados
por especialistas en hipertensión experimentados. La cirugía es el tratamiento
de elección y puede ser un procedimiento de alto riesgo. La preparación
preoperatoria generalmente combina bloqueadores alfa (como fenoxibenzamina,
ajustado a la hipotensión postural) con bloqueadores de los canales de calcio o
beta, según sea necesario. Algunos médicos recomiendan metirosina, pero la
experiencia con este inhibidor de la síntesis de catecolaminas es limitada.
Las crisis perioperatorias se manejan con
nitroprusiato. La resección quirúrgica es prudente para los feocromocitomas
malignos, aunque la supervivencia a largo plazo es escasa.
SÍNDROME DE CUSHING
Los niveles de glucocorticoides crónicamente elevados
dan como resultado síntomas y signos proteicos que son comunes (como obesidad,
hiperglucemia e hipertensión) e inespecíficos (debilidad, acné, edema, estrías,
dolor de cabeza, plétora). Estas características hacen que el diagnóstico del
síndrome de Cushing sea un desafío. La hipercortisolemia del síndrome de
Cushing puede originarse en las suprarrenales o ser el resultado de un adenoma
hipofisario u otros tumores secretores de ACTH.
CARACTERÍSTICAS CLÍNICAS
La característica de presentación más común es la
obesidad de la cara, el cuello y el abdomen que respeta las extremidades. El
depósito de grasa facial puede dar lugar a una cara redondeada, exacerbada por
el depósito de grasa en las almohadillas de grasa supraclaviculares, lo que
hace que el cuello parezca más corto. Estos pacientes desarrollan
adelgazamiento de la piel, atrofia y moretones con facilidad. Las estrías
típicas del síndrome de Cushing suelen ser de color púrpura, anchas y
múltiples, factores que ayudan a distinguirlas de las estrías asociadas con la
obesidad. Las mujeres con enfermedad de Cushing pueden tener signos de
hiperandrogenismo, como hirsutismo. La miopatía proximal (generalmente descrita
como dificultad para levantarse de una posición sentada), cambios psiquiátricos
(labilidad emocional, depresión y paranoia leve son comunes) e hipertensión a
menudo están presentes en la presentación. Otras características asociadas con
una hipercortisolemia más prolongada o más grave incluyen intolerancia a la
glucosa, glaucoma y osteopenia.
EVALUACIÓN
El primer paso para evaluar si una paciente puede
tener síndrome de Cushing es dilucidar cualquier antecedente de exposición a
corticosteroides, incluidos esteroides potentes inhalados, inyectados o
tópicos, o acetato de medroxiprogesterona (una progestina con actividad
esteroidea intrínseca). El síndrome de Cushing facticio representa <1% de
todos los casos y se sugiere por resultados erráticos e inconsistentes. En
tales casos, los glucocorticoides sintéticos pueden analizarse directamente en
la orina.
Para establecer hipercortisolemia, se deben obtener al
menos dos muestras de orina de 24 horas para cortisol libre (y creatinina). Se
puede asumir que los pacientes cuyos niveles son más de tres veces más altos
que el rango de referencia superior tienen síndrome de Cushing. Los pacientes
con valores equívocos deben volver a analizarse después de algunas semanas o
evaluarse con más pruebas según la sospecha clínica.
Una prueba de supresión con dexametasona durante la
noche también se usa como prueba de detección para diagnosticar la
hipercortisolemia. En esta prueba, se extrae un nivel de cortisol sérico a las
8 am después de una dosis de 1 mg de dexametasona entre las 11 pm y la
medianoche. La mayoría de los pacientes normales deben suprimir su nivel de
cortisol endógeno a <2 μg/dl. Una estrategia de detección que utiliza tres
cortisoles salivales consecutivos al final de la noche puede reemplazar en
última instancia a estas pruebas antes mencionadas. Estos pueden ser realizados
por un paciente ambulatorio con instrucción mínima. Los rangos de referencia
son específicos del laboratorio.
Una vez que el diagnóstico de síndrome de Cushing es
seguro, debe buscarse una fuente para la hipercortisolemia. Determinar si el
síndrome de Cushing es dependiente de ACTH o de origen suprarrenal requiere una
medición precisa de los niveles de ACTH. Se puede considerar que la secreción
de cortisol es independiente de ACTH si la ACTH es <5 pg/mL cuando el nivel
de cortisol es >15 μg/dL. En las mismas circunstancias, es muy probable que
el síndrome sea dependiente de ACTH si la concentración de ACTH es >15 pg/mL
cuando el cortisol es de al menos 15 μg/dL; Los niveles de ACTH entre 5 y 15
pg/mL son menos específicos pero generalmente indican dependencia de ACTH. Los
pacientes con valores equívocos deben ser investigados nuevamente.
Los pacientes con hipercortisolemia independiente de ACTH
deben someterse a una TC de corte fino de las glándulas suprarrenales para
identificar el adenoma, carcinoma o nódulos responsables.
Los pacientes con hipercortisolemia dependiente de
ACTH deben someterse a pruebas adicionales para discriminar entre la enfermedad
de Cushing relacionada con un adenoma hipofisario y la relacionada con la
secreción ectópica de ACTH. Los tumores reconocidos por secretar ACTH incluyen
el cáncer de células pequeñas del pulmón y los carcinoides bronquiales y
tímicos. Los médicos deben resistir la tentación de obtener imágenes de la
pituitaria porque el 10% de la población tiene una pituitaria estructuralmente
anormal. En su lugar, los pacientes deben someterse a una prueba de supresión
con dosis altas de dexametasona, ya sea con 2 mg de dexametasona administrados
cada 6 horas durante 2 días o con 8 mg de dexametasona administrados entre las
11 p. m. y la medianoche, antes de la medición matutina de cortisol. Esta
prueba aprovecha el hecho de que los adenomas hipofisarios secretores de ACTH
retienen cierta capacidad de respuesta a la retroalimentación y, a menudo,
suprimen su producción de ACTH cuando los niveles ambientales de
glucocorticoides son altos. Los niveles de cortisol se reducen en >90% entre
el 70% de las personas con enfermedad de Cushing. En el mismo estudio, por
contraste, ningún paciente con ACTH de origen ectópico suprimió el cortisol por
debajo del 90% en respuesta a esta prueba de supresión de dosis alta.
El muestreo de los senos paranasales mediante la
estimulación de la hormona liberadora de corticotropina (CRH) es un enfoque
final para confirmar que la ACTH se deriva de la hipófisis. Los criterios para
confirmar que la hipófisis es la fuente de la ACTH incluyen una relación de
ACTH entre un lado del seno petroso y el plasma periférico de >2 o una
relación >3 durante la infusión de CRH en comparación con el nivel antes de
que se inicie la infusión. Si un lado tiene un nivel de ACTH que es un múltiplo
de 1,4 veces o más que el nivel del lado opuesto, es muy probable que el
adenoma resida en ese lado. A los pacientes con sospecha de ACTH ectópica se
les debe realizar una imagen con octreótido con imágenes torácicas simples y
tomográficas obtenidas según se indica.
TRATAMIENTO
El objetivo del tratamiento del síndrome de Cushing es
la erradicación de cualquier tumor, la supresión de los niveles de cortisol al
mínimo posible y evitar la dependencia hormonal permanente. El tratamiento de
elección para la enfermedad de Cushing es la resección hipofisaria
transesfenoidal, independientemente del tamaño del tumor hipofisario. Cuanto
más extensa es la resección, mayor es el riesgo de hipopituitarismo permanente.
Esto puede tener implicaciones particulares para los pacientes más jóvenes que
aún no han formado una familia. La radiación hipofisaria se puede administrar a
pacientes con tumores irresecables o residuales, aunque esto se asocia con una
alta tasa de hipopituitarismo.
Los inhibidores de la esteroidogénesis, los
antagonistas de los receptores de glucocorticoides o las terapias médicas
dirigidas a la hipófisis son opciones adicionales para los pacientes con
hipercortisolemia persistente.
Los tumores suprarrenales que causan hipercortisolemia
se extirpan mejor. El tratamiento médico de los tumores irresecables o de los
pacientes con cáncer suprarrenal hormonalmente activo metastásico es un desafío
porque estas neoplasias malignas responden poco a las terapias adyuvantes. Los
pacientes pueden beneficiarse del uso de mitotano, un veneno suprarrenal. Estos
pacientes deben recibir glucocorticoides suplementarios en dosis de reemplazo
para garantizar que no desarrollen insuficiencia suprarrenal durante el
tratamiento. Los pacientes con hipercortisolemia incontrolable pueden
beneficiarse de los inhibidores de las enzimas esteroides suprarrenales como el
ketoconazol o la metirapona. La quimiorradioterapia experimental o agentes
adicionales pueden estar disponibles como parte de un ensayo clínico.
HIRSUTISMO
El hirsutismo se refiere a la aparición de vello
terminal excesivo que aparece en un patrón masculino en las mujeres.
Aproximadamente el 5% de las mujeres son hirsutas. El hirsutismo resulta de una
interacción entre el nivel de andrógenos y la sensibilidad del folículo piloso
a los andrógenos; como resultado, los niveles de andrógenos no se correlacionan
bien con el grado de hirsutismo. Aproximadamente la mitad de las mujeres con
hirsutismo tienen la condición idiopática.
CARACTERÍSTICAS CLÍNICAS
Las características clínicas que sugieren una de las
causas raras o más graves de hirsutismo incluyen un inicio repentino, una
presentación más tardía en la vida y un empeoramiento progresivo. Los síntomas
y signos de virilización incluyen calvicie frontal, acné, clitoromegalia y voz
grave. Debe evaluarse el crecimiento de vello en el labio superior, el mentón,
el pecho, el abdomen, la espalda, el pubis y las piernas. El hirsutismo debe
distinguirse de la hipertricosis, la aparición de un crecimiento de vello
excesivo generalizado determinado genéticamente o que sigue al tratamiento con glucocorticoides,
fenitoína o ciclosporina.
EVALUACIÓN
Si el hirsutismo es moderado o severo, la testosterona
plasmática y la testosterona libre deben medirse temprano en la mañana
(idealmente en los días 4 a 10 del ciclo menstrual en mujeres premenopáusicas).
El hiperandrogenismo se relaciona con mayor frecuencia
con el síndrome de ovario poliquístico, uno de los trastornos hormonales más
comunes que afectan a las mujeres. El síndrome se diagnostica cuando la
paciente tiene al menos dos de hiperandrogenismo crónico, oligoovulación o
anovulación y ovarios poliquísticos, y se excluyen otros diagnósticos. Estos
pacientes a menudo tienen irregularidad menstrual, obesidad y evidencia de
resistencia a la insulina (p. ej., acantosis nigricans). No se requiere una ecografía
pélvica para el diagnóstico. Las pruebas adicionales pueden incluir una prueba
de embarazo si la paciente tiene amenorrea. Estos pacientes deben ser evaluados
por intolerancia a la glucosa y apnea del sueño y, a menudo, responden bien a
los sensibilizadores de insulina como la metformina o una tiazolidinediona. La
espironolactona y los anticonceptivos orales se usan con frecuencia para
controlar el hirsutismo en estos pacientes.
Otras causas de hiperandrogenismo son inusuales. La
hiperplasia suprarrenal congénita virilizante es sugerida por el crecimiento
prematuro del vello púbico y la clitoromegalia y puede excluirse midiendo el
nivel matutino de 17-alfa-hidroxiprogesterona.
El síndrome de Cushing se sugiere por el desarrollo de
obesidad troncal, cara de luna llena, joroba de búfalo, estrías moradas o
debilidad muscular proximal. La hiperprolactinemia se sugiere por la presencia
de galactorrea y un nivel elevado de prolactina. La acromegalia se sugiere por
el engrosamiento de los rasgos faciales o por el agrandamiento de las manos y
se confirma por un nivel elevado de IGF-1.
Los tumores secretores de andrógenos son muy raros,
pero deben considerarse entre mujeres con una presentación aguda o que tienen
niveles muy altos de testosterona (>200 ng/dL). Estas mujeres deben ser
evaluadas con un nivel de sulfato de dehidroepiandrosterona y una imagen
abdominal y pélvica.
El hirsutismo idiopático es el diagnóstico más común
después de que estos otros trastornos hayan sido excluidos por características clínicas
o de laboratorio.
TRATAMIENTO
El hirsutismo se puede controlar con terapia cosmética
y hormonal. Es útil completar una evaluación objetiva del grado de hirsutismo
antes de iniciar el tratamiento. La puntuación de Ferriman-Gallwey es uno de
esos sistemas de puntuación.
Los enfoques cosméticos incluyen la decoloración, el
afeitado, la cera, la electrólisis, el tratamiento con láser y el uso de
agentes depilatorios. La crema de clorhidrato de elornitina se puede usar para
el hirsutismo facial, pero debe usarse aproximadamente 8 semanas antes de que
se pueda determinar su eficacia.
Los anticonceptivos de estrógeno-progestina reducen
los niveles de andrógenos circulantes y pueden reducir la necesidad de
afeitarse y retrasar la progresión del hirsutismo. Se prefieren los
anticonceptivos con progestágenos no androgénicos. Se pueden ofrecer
antiandrógenos cuando el hirsutismo es de moderado a severo. La espironolactona
en dosis altas (50 a 100 mg dos veces al día) es eficaz para reducir el
hirsutismo. Se debe informar a los pacientes que la espironolactona puede ser
teratogénica y, por lo general, no se prescribe a mujeres sexualmente activas
sin el uso concomitante de un anticonceptivo oral. La hiperpotasemia rara vez
se asocia con espironolactona entre mujeres con función renal normal. La
flutamida es un antiandrógeno que se asocia con hepatotoxicidad y generalmente
no se recomienda para controlar el hirsutismo. El acetato de ciproterona es un
antiandrógeno disponible en Canadá, México y Europa, pero no en los Estados
Unidos.
HIPOGONADISMO MASCULINO
La deficiencia de testosterona puede resultar de una
enfermedad de los testículos, o de una disfunción pituitaria o hipotalámica. Estas
causas se pueden distinguir midiendo las gonadotropinas, la hormona
luteinizante y la hormona estimulante del folículo.
CARACTERÍSTICAS CLÍNICAS
La deficiencia de testosterona intrauterina puede resultar en micropene y criptorquidia y, cuando ocurre antes de la pubertad, la deficiencia de testosterona resultará en una maduración incompleta, un hábito eunucoide y una masa ósea máxima reducida. Cuando la deficiencia de testosterona ocurre después de la pubertad, se produce una disminución de la libido y de la función eréctil, con pérdida de vello sexual, masa muscular y densidad mineral ósea. La ginecomastia puede estar presente.
EVALUACIÓN
Las causas de la testosterona baja deben buscarse en
la historia, incluidas las indicaciones de consumo de opiáceos, apnea del
sueño, hemocromatosis, traumatismo testicular, orquitis y obesidad. El testículo
adulto normal mide aproximadamente de 3,5 a 5,5 cm de largo o de 15 a 30 ml de
volumen. La extensión de los brazos normalmente no debe ser más de 5 cm más
larga que la altura. El agrandamiento de las mamas, los testículos pequeños y las
anomalías del comportamiento sugieren síndrome de Klinefelter. La anosmia
sugiere síndrome de Kallmann.
Las concentraciones de testosterona total y de
globulina transportadora de hormonas sexuales deben medirse en una muestra
matutina con hormona luteinizante y hormona estimulante del folículo. Las
mediciones de testosterona libre generalmente no son confiables. El análisis de
semen es apropiado si la infertilidad es una preocupación principal.
TRATAMIENTO
El tratamiento con testosterona debe reservarse para
hombres con síntomas clínicos y signos de hipogonadismo acompañados de una
concentración de testosterona por debajo de lo normal que tienen un nivel de
antígeno prostático específico normal. Los ésteres de testosterona se pueden
administrar por inyección intramuscular semanal o quincenalmente. Los geles
transdérmicos de testosterona están disponibles en sobres y una bomba
dosificadora y se administran diariamente después de la ducha; los pacientes
deben tener cuidado con la transmisión de piel a piel a un compañero de cama.
Los parches de testosterona están disponibles pero causan irritación local. La
dosis se titula a un nivel de testosterona total matutino, tomado antes de la
siguiente dosis. Los médicos deben controlar los síntomas de hipertrofia
prostática benigna, apnea del sueño y acné, y controlar el antígeno prostático
específico y el hematocrito para detectar eritrocitosis.
DIABETES MELLITUS
La prevalencia de la diabetes mellitus está aumentando
exponencialmente en todo el mundo. La enfermedad afecta aproximadamente a 20
millones de estadounidenses, y muchos más aún no se han diagnosticado. La
diabetes tipo 2 representa aproximadamente el 95% de todos los casos y se
caracteriza por resistencia a la insulina e hiperglucemia. La hiperinsulinemia
ocurre en las primeras etapas de la enfermedad, pero no se mantiene
indefinidamente. Muchos pacientes finalmente requieren insulina para mantener
la glucosa en el rango normal. Debido a que casi el 80% de todos los pacientes
con diabetes morirán por complicaciones cardiovasculares, la reducción del
riesgo cardiovascular es el objetivo principal.
CARACTERÍSTICAS CLÍNICAS
La diabetes se diagnostica si hay síntomas de diabetes
(poliuria, polidipsia, pérdida de peso inexplicable) y una glucosa aleatoria de
≥200 mg/dl. Una glucosa en ayunas ≥126 mg/dl o una glucosa ≥200 mg/dl 2 horas
después de una carga de glucosa de 75 g también es diagnóstica. Una hemoglobina
A1c (HbA1c) de al menos 6,5% también es diagnóstica. Se recomienda la detección
de diabetes cada 3 años para todos los pacientes con sobrepeso con factores de
riesgo y en todos a partir de los 45 años.
La prediabetes está indicada por una HbA1c entre 5.7%
y 6.5%, alteración de la glucosa en ayunas (glucosa en ayunas 100 a 125 mg/dl)
o alteración de la tolerancia a la glucosa (glucosa 140 a 199 mg/dl 2 horas
después de una carga de glucosa de 75 g). La incidencia de diabetes se puede
reducir en más del 50 % en pacientes con prediabetes si los pacientes pierden
peso y se embarcan en un programa de ejercicio que comprende al menos 30
minutos de ejercicio cinco veces por semana. La metformina reduce la incidencia
de diabetes en estos pacientes de alto riesgo en aproximadamente un 25%.
EVALUACIÓN
Se deben considerar las causas secundarias de la
diabetes al evaluar a cualquier paciente con hiperglucemia recién
diagnosticada. Los medicamentos que están asociados con la hiperglucemia, como
los glucocorticoides, los antipsicóticos y algunos antirretrovirales, deben
reevaluarse para determinar si un agente alternativo puede sustituirse de manera
segura. Las causas genéticas de la diabetes deben excluirse si hay antecedentes
familiares importantes de diabetes o si se observa un fenotipo típico (p. ej.,
síndrome de Down, síndrome de Turner o síndrome de Klinefelter). Las
endocrinopatías como el síndrome de Cushing, la acromegalia, el feocromocitoma,
el hipertiroidismo y otras deben buscarse a partir de la historia clínica y el
examen. Los pacientes con enfermedades que afectan al páncreas exocrino, como
la hemocromatosis, la pancreatitis crónica, las neoplasias malignas del
páncreas o la fibrosis quística, tienen un alto riesgo de diabetes; el
tratamiento de la enfermedad subyacente suele ser fundamental para reducir la
tasa de progresión a la deficiencia de insulina y controlar la hiperglucemia.
TRATAMIENTO
A los pacientes a los que se les acaba de diagnosticar
diabetes tipo 2 se les debe proporcionar un glucómetro e instrucciones para las
pruebas, y se les debe derivar para recibir educación sobre la diabetes y
terapia de nutrición médica. no se ha determinado la frecuencia óptima de las
pruebas de glucosa por punción en el dedo. Se debe enfatizar el abandono del
hábito de fumar y los beneficios del ejercicio y la pérdida de peso. Los
objetivos del tratamiento se enumeran en la Tabla 57.3.

Tabla 57.3. Metas de tratamiento para pacientes con
diabetes
Para los pacientes con diabetes tipo 2 que requieren
tratamiento, la metformina sigue siendo el agente de elección de primera línea.
Se debe advertir a los pacientes que los efectos secundarios gastrointestinales
tempranos no son infrecuentes y deben tolerarse si es posible; estos
generalmente abad dentro de 2 semanas. El uso de metformina está asociado con
la reducción del riesgo cardiovascular, pero debe usarse con precaución en
pacientes cuya creatinina > 1,5 mg/dl o entre aquellos que tienen
enfermedades crónicas graves.
Las sulfonilureas generalmente se recomiendan como
agentes de segunda línea debido a su alta eficacia y bajo costo. Se prefieren
las sulfonilureas de acción corta como la glipizida por su vida media más
corta, especialmente entre los pacientes de edad avanzada. Las meglitinidas
relacionadas son menos potentes y más costosas.
Los enfoques más recientes para el control de la
glucemia incluyen el uso de análogos del péptido similar al glucagón (GLP)-1 e
inhibidores de la dipeptidil-peptidasa-4 (DPP-4). Los análogos de GLP-1, como
exenatida, liraglutida y dulaglutida, proporcionan mejoras modestas en la HbA1c
y pueden contribuir a la pérdida de peso en algunos pacientes; a menudo se usan
en lugar de la insulina en aquellos que están cerca de su objetivo de HbA1c y
que pueden beneficiarse de la pérdida de peso.
Los inhibidores de DPP-4 como sitagliptina,
linagliptina, alogliptina y saxagliptina parecen ser seguros y bien tolerados,
pero su eficacia se limita a una caída de HbA1c de <0,7 puntos porcentuales.
Los inhibidores del transporte de sodio y glucosa,
como la canaglifozina, la dapaglifozina y la empaglifozina, aumentan la
glucosuria y parecen disminuir la glucosa y la presión arterial de forma
dependiente de la glucosa, lo que reduce el riesgo de hipoglucemia. Se asocian
con un aumento de infecciones pélvicas menores.
A pesar de su asociación con una presión arterial más
baja y la ausencia de hipoglucemia, la tiazolidinediona pioglitazona rara vez
se recomienda porque causa retención de líquidos, aumento de peso y pérdida
ósea prematura.
Para los pacientes con diabetes tipo 2 que permanecen
hiperglucémicos a pesar de dos agentes hipoglucemiantes, la insulina es una
opción adecuada. Una dosis inicial típica de insulina es de 10 a 20 U. El
tratamiento con insulina debe incluir una insulina basal (como insulina
glargina, detemir o isófana [NPH]) ajustada inicialmente a una glucosa en
ayunas de aproximadamente 100 mg/dL. Si la HbA1c permanece elevada, se puede
agregar gradualmente una insulina de acción corta (como insulina aspart,
glulisina o lispro), comenzando con una dosis antes de la comida principal y
luego antes de otras comidas. La dosis de insulina de acción corta se puede
ajustar a un nivel de glucosa posprandial tomado aproximadamente 2 a 3 horas
después de una comida de prueba. Se debe recomendar a los pacientes que
necesitan tratamiento con insulina que verifiquen sus niveles de glucosa antes
de conducir.
Los pacientes con diabetes tipo 1 requieren
tratamiento con insulina de por vida. Por lo general, la insulina basal se usa
con insulinas de acción ultracorta que se administran antes de cada comida o
refrigerio. A los pacientes con diabetes tipo 1 se les puede enseñar a contar los
carbohidratos y cómo calcular la dosificación de insulina prandial y de
corrección. Estos pacientes deben trabajar con un equipo de diabetes y pueden
beneficiarse de la conveniencia de la terapia con bomba de insulina,
especialmente cuando se combina con un control continuo de glucosa. Las pautas
para el cuidado de la presión arterial, los lípidos y los riñones, los ojos y
los pies son similares a las de los pacientes con diabetes tipo 2.
PREVENCIÓN DE COMPLICACIONES
El control de la presión arterial es al menos tan
importante como el control de la glucemia para reducir la incidncia de
enfermedades cardiovasculares a largo plazo en pacientes con diabetes. El
tratamiento con un inhibidor de la ECA o un bloqueador de los receptores de la
angiotensina generalmente se usa como primera línea, y se agregan diuréticos
tiazídicos o bloqueadores de los canales de calcio según sea necesario.
Los niveles de microalbúmina urinaria deben evaluarse
anualmente. La presencia de >30 mg de microalbúmina por gramo de creatinina
es un factor de riesgo de nefropatía y enfermedad cardiovascular. Los
inhibidores de la ECA o los bloqueadores de los receptores de angiotensina
deben iniciarse en pacientes con diabetes y microalbuminuria para disminuir el
riesgo de insuficiencia renal progresiva.
Las estatinas deben ofrecerse a pacientes con diabetes
y mayores de 40 años. La terapia con estatinas de alta intensidad se recomienda
para pacientes con enfermedad cardiovascular aterosclerótica (ASCV) o aquellos
entre 40 y 75 años de edad con factores de riesgo cardiovascular adicionales.
Se debe considerar la adición de estatinas de intensidad moderada para
pacientes entre 40 y 75 años y sin factores de riesgo de ASCV. Se puede
considerar la terapia con estatinas de intensidad moderada o alta en pacientes
> 75 años de edad o < 40 años con factores de riesgo adicionales de ASCV.
Agregar ezetimiba a la terapia con estatinas de intensidad moderada puede
proporcionar un beneficio cardiovascular incremental para aquellos que no
pueden tolerar las estatinas de alta intensidad. La hiperlipidemia en la
diabetes típicamente presenta hipertrigliceridemia y niveles bajos de
lipoproteínas de alta densidad. Aunque este último puede tratarse con ibratos o
niacina, hay poca evidencia de que este tratamiento reduzca el riesgo
cardiovascular.
DIABETES GESTACIONAL
La prevalencia de la diabetes en el embarazo ha ido en
aumento debido al aumento tanto de la diabetes gestacional (DMG) como de la
diabetes pregestacional. La diabetes pregestacional confiere mayor riesgo de
complicaciones maternas y fetales que la DMG. El aumento exponencial de la
resistencia a la insulina durante el segundo y tercer trimestre es un factor
importante de DMG.
Las mujeres con factores de riesgo de diabetes deben
someterse a un examen de detección de diabetes antes de la concepción o durante
la primera visita prenatal utilizando criterios de diagnóstico estándar. Las
mujeres con diabetes en el primer trimestre tienen diabetes manifiesta, no DMG.
La detección de GDM se realiza de manera óptima entre las 24 y 28 semanas de
gestación.
El diagnóstico de diabetes gestacional durante el
embarazo se puede realizar en mujeres que tienen una glucosa plasmática en
ayunas de al menos 92 mg/dl o una glucosa superior a 180 mg/dl 1 hora o
superior a 153 mg/dl 2 horas después una carga de glucosa de 75 g. Existen
diferentes criterios para el diagnóstico utilizando un enfoque de dos pasos con
una carga de glucosa de 50 g seguida de 100 g.
Las pacientes con diabetes gestacional deben recibir
asesoramiento nutricional y un glucómetro. Las lecturas de glucosa en ayunas
deben mantenerse ≤95 mg/dL y los niveles posprandiales deben ser ≤140 mg/dL a
la hora y ≤120 mg/dL a las 2 horas. El objetivo de HbA1c debe ser <6% a
6,5%. Una minoría de mujeres con DMG requerirá tratamiento con insulina;
normalmente se utilizan insulina NPH e insulinas de acción ultracorta (lispro o
aspart). La metformina se puede usar para controlar la diabetes gestacional,
pero se debe informar a los pacientes que se desconocen los efectos a largo
plazo.
HIPERGLUCEMIA EN EL HOSPITAL
La hiperglucemia durante la hospitalización se asocia
con aumentos significativos de la morbilidad y la mortalidad. En entornos de
cuidados intensivos, las infusiones intravenosas de insulina a menudo se recomiendan
si la glucosa es >180 mg/dL y luego se titula a un objetivo de glucosa de
140 a 180 mg/dL. Los objetivos más bajos pueden ser apropiados para pacientes
seleccionados. El paciente puede hacer la transición a insulina subcutánea
cuando esté estable (p. ej., extubado, con vasopresores) ya sea que esté
comiendo o no. Si el control de la glucosa ha sido el objetivo, muchos médicos
comienzan con una dosis de insulina que es el 80 % del uso diario total de
insulina del día anterior. Las recetas deben escribirse para dosis basales,
prandiales y de corrección.
SÍNDROMES ENDOCRINOS
NEOPLASIA ENDOCRINA MÚLTIPLE
Los tipos 1, 2A y 2B de MEN son síndromes genéticos
raros que comprenden múltiples tumores hormonalmente activos y algunos tipos de
cáncer (tabla 57.4).
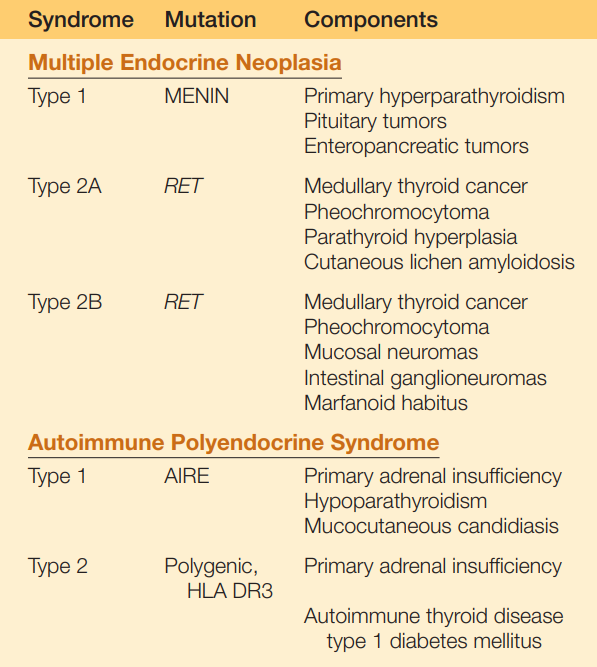
Tabla 57.4. Componentes de la Neoplasia Endocrina
Múltiple y Síndromes Poliendocrinos Autoinmunes
NEOPLASIA ENDOCRINA MÚLTIPLE 1
El hiperparatiroidismo es la manifestación más común
de MEN-1, causada por hiperplasia de múltiples glándulas paratiroides. La
penetrancia es casi del 100% a la edad de 50 años. Los segundos tumores más
comunes son los tumores pancreáticos productores de polipéptido pancreático.
Los gastrinomas ocurren en aproximadamente el 60% de los pacientes con MEN-1 y,
a menudo, son metastásicos en el momento del diagnóstico. Los glucagonomas son
raros. Los prolactinomas son los tumores hipofisarios más comunes en pacientes
con MEN-1, pero la acromegalia ocurre en alrededor del 25% de los pacientes.
Otros tumores que se informa que ocurren en pacientes con el síndrome MEN-1
incluyen tumores carcinoides del intestino anterior, angiobromas, lipomas y
adenomas suprarrenales benignos.
NEOPLASIA ENDOCRINA MÚLTIPLE 2
El MEN tipo 2A representa casi el 95% de los casos de
MEN tipo 2. La hiperplasia de células C es un precursor del cáncer medular de
tiroides que surge de forma multifocal y bilateral. Los feocromocitomas ocurren
entre la tercera y la cuarta década de la vida y suelen ser bilaterales. El
hiperparatiroidismo franco es inusual. Los pacientes con sospecha de MEN-2
deben someterse a pruebas para la mutación de la línea germinal RET y exámenes
de detección anuales con metanefrinas en plasma. Se recomienda la tiroidectomía
profiláctica con disección de ganglios linfáticos en niños menores de 5 años
que tienen una mutación de la línea germinal RET en el exón 16.
SÍNDROME POLIENDOCRINO AUTOINMUNE
Los síndromes autoinmunes poliglandulares son
constelaciones de múltiples insuficiencias de las glándulas endocrinas. Estos
son trastornos extremadamente raros y generalmente son evidentes desde la
adolescencia temprana.
El orden de aparición de los componentes del síndrome
poliendocrino autoinmune tipo 1 es generalmente candidiasis, hipoparatiroidismo
y luego insuficiencia suprarrenal. El panel de detección de anticuerpos puede
incluir autoanticuerpos contra 21-hidroxilasa, 17-hidroxilasa, peroxidasa
tiroidea e inmunoglobulinas estimulantes de la tiroides, ácido glutámico
descarboxilasa y anticuerpos contra células de los islotes, y anticuerpos
contra enzimas de células parietales.
El síndrome poliendocrino autoinmune tipo 2 es más
común que el tipo 1. La insuficiencia suprarrenal primaria es un componente
obligatorio. El hipogonadismo primario, la celiaquía y la miastenia grave
también pueden complicar la presentación. El inicio es generalmente en la
cuarta década o más tarde, con predominio femenino.
FUENTE:
The
Brigham Intensive Review of
Internal Medicine. (2022)
Ajay K. Singh, MBBS, FRCP, MBA
Joseph Loscalzo, MD, PhD
No hay comentarios:
Publicar un comentario