Un hombre de 78 años fue evaluado en la consulta de cardiología de este hospital por engrosamiento de la pared miocárdica biventricular, insuficiencia mitral progresiva y arritmias ventriculares.
El paciente había sido un distinguido atleta
multideportivo en la escuela secundaria y la universidad. Sin embargo, cuando
tenía 20 años, descubrió que no podía desempeñarse de manera competitiva y notó
que no sudaba tanto como sus compañeros de equipo. Después de un episodio de
mareo por esfuerzo relacionado con el deporte, fue evaluado por un médico y se
consideró un diagnóstico de miocardiopatía hipertrófica.
Veinticinco años antes de la evaluación actual, se
informó que un ecocardiograma transtorácico (ETT) mostró un grosor simétrico de
la pared del ventrículo izquierdo de 19 mm (valor de referencia, ≤11), una
fracción de eyección del ventrículo izquierdo del 74 % (rango de referencia, 50
a 75), engrosamiento de la válvula mitral y rastro de insuficiencia mitral, sin
gradiente de salida del ventrículo izquierdo. Un electrocardiograma mostró
ritmo sinusal, bloqueo incompleto de rama derecha, elevación del punto J
precordial y voltaje QRS que cumplía con los criterios electrocardiográficos de
hipertrofia ventricular izquierda ( Figura 1A ). La presión arterial del
paciente era de 160/90 mm Hg, y comenzó tratamiento con succinato de metoprolol
y luego también con amlodipino por hipertensión arterial.

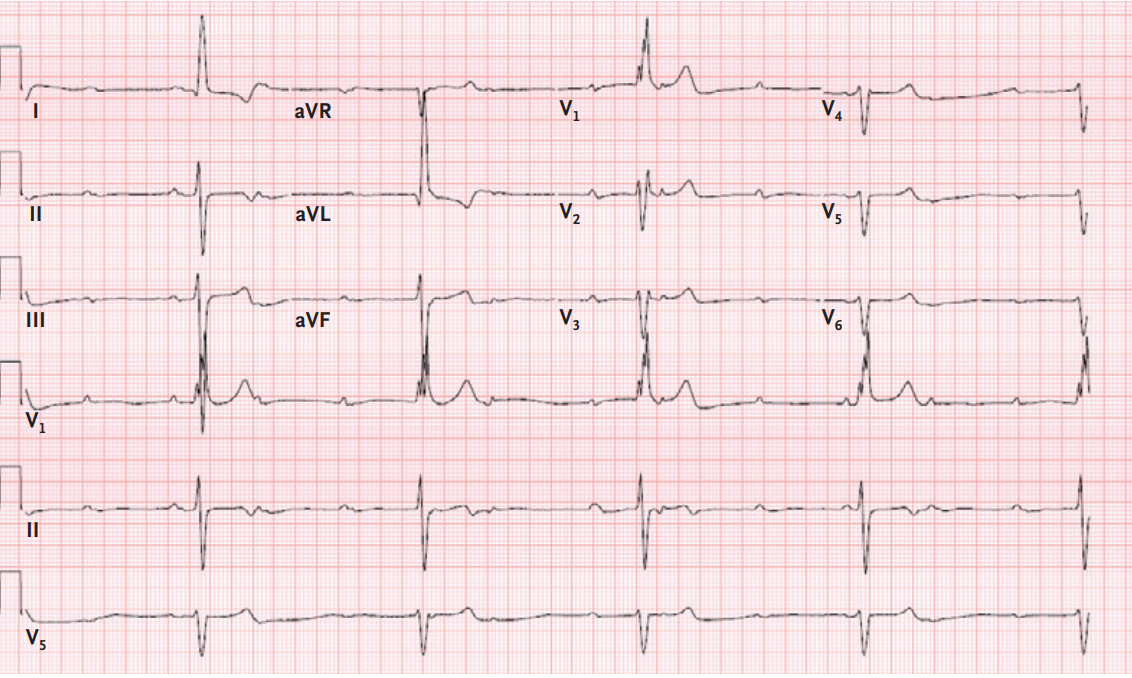
Figura 1. Electrocardiogramas.
Un electrocardiograma obtenido 25 años antes de la
evaluación actual (Panel A) muestra ritmo sinusal, con bloqueo incompleto de
rama derecha, elevación del punto J precordial y voltaje QRS que cumple con los
criterios electrocardiográficos para hipertrofia ventricular izquierda. Los
trazados electrocardiográficos obtenidos durante los siguientes 15 años
mostraron hallazgos similares. Un electrocardiograma obtenido 9 años antes de
la evaluación actual (Panel B) muestra ritmo sinusal con bloqueo cardíaco
completo y ritmo de escape con bloqueo de rama derecha y desviación del eje a
la izquierda.
Durante los siguientes 10 años, el paciente tuvo
molestias torácicas intermitentes, disnea de esfuerzo y edema en las piernas.
Las molestias torácicas y la disnea de esfuerzo ocurrían con mayor probabilidad
cuando caminaba o después de haber comido. Fue evaluado por tres cardiólogos;
Se continuó tratamiento con succinato de metoprolol y amlodipino, se inició
tratamiento con enalapril y se administró triamtereno-hidroclorotiazida a
demanda por edema de piernas.
Según los informes, un ETT repetido mostró un grosor
simétrico de la pared del ventrículo izquierdo de 20 mm, una fracción de
eyección del ventrículo izquierdo normal, engrosamiento de la pared del
ventrículo derecho, agrandamiento de la aurícula izquierda, engrosamiento de la
válvula mitral, regurgitación mitral de leve a moderada, engrosamiento de la válvula
aórtica y leve dilatación de la aorta ascendente (39 mm), sin evidencia de
obstrucción del tracto de salida del ventrículo izquierdo y sin evidencia de
movimiento anterior sistólico de la válvula mitral. Una prueba de esfuerzo con
ejercicio seguida de imágenes realizadas con el uso de sestamibi (conocida como
prueba de esfuerzo con sestamibi) reveló un engrosamiento de la pared del
ventrículo izquierdo, sin defectos de perfusión. La angiografía coronaria
reveló evidencia de oclusión total crónica de la arteria descendente anterior
izquierda distal, con flujo colateral de las arterias coronarias izquierda y
derecha. La presión telediastólica del ventrículo izquierdo era de 28 mmHg
(rango de referencia, 5 a 10), sin gradiente intracavitario del ventrículo
izquierdo, incluso durante los latidos ectópicos ventriculares y durante la
maniobra de Valsalva, y sin evidencia de obstrucción del tracto de salida del
ventrículo izquierdo. La presión arterial se mantuvo bien controlada mientras
el paciente tomaba metoprolol, amlodipino y enalapril.
Nueve años antes de la evaluación actual, el paciente
tenía un bloqueo cardíaco completo con bradicardia sintomática ( Figura 1B),
con náuseas, mareos y malestar torácico asociados. Se colocó un cable de
marcapasos transvenoso temporal; los resultados de la angiografía coronaria
realizada en ese momento no cambiaron con respecto al estudio anterior. El
nivel de creatinina fue de 1,2 mg por decilitro ( 0,6 a 1,5 mg por decilitro);
los niveles sanguíneos de electrolitos y tirotropina eran normales, al igual
que los resultados de la electroforesis de proteínas séricas. Se colocó un
desfibrilador cardioversor implantable (CDI) bicameral. Cuatro meses después,
desarrolló fibrilación auricular. Se realizó cardioversión guiada por
ecocardiografía transesofágica, resultando en ritmo sinusal. Sin embargo, la
fibrilación auricular reapareció después de 3 días. Se inició tratamiento con
amiodarona y warfarina.
Durante los siguientes 5 años, se detectaron
taquiarritmias ventriculares no sostenidas recurrentes en el interrogatorio del
CDI. Un ETT repetidO mostró un grosor de la pared del ventrículo izquierdo de
23 mm e insuficiencia mitral de moderada a grave.
Cuatro años antes de la evaluación actual, durante un
viaje de vacaciones, el paciente fue hospitalizado en otro estado debido a un
edema pulmonar que se desarrolló después de haber consumido alimentos con alto
contenido de sodio. Fue tratado con furosemida endovenosa y dado de alta con
prescripción de furosemida oral. Según los informes, los resultados de la ETT
fueron similares a los observados anteriormente. Se suspendió el tratamiento
con amlodipina debido a mareos.
En las citas de seguimiento durante los siguientes 4
años, el paciente informó fatiga y disnea de esfuerzo que ocurrieron después de
haber subido un tramo de escaleras o cuando caminaba en una pendiente. Además,
tenía nicturia una vez por noche, dormía sobre dos almohadas y tenía
sibilancias intermitentes, incluidos episodios que lo despertaban del sueño. Se
continuó el tratamiento con furosemida oral (administrada dos veces al día). Se
detectaron taquiarritmias ventriculares recurrentes no sostenidas en el
interrogatorio del CDI. Aproximadamente 3,5 años antes de la evaluación actual,
el paciente sufrió una caída que no estaba relacionada con su afección
cardíaca. En ese momento, la tomografía computarizada de la cabeza, realizada
sin la administración de material de contraste intravenoso, no reveló ninguna
evidencia de hemorragia o accidente cerebrovascular, pero mostró hipodensidades
periventriculares y profundas de la sustancia blanca. Los resultados de las
pruebas de función pulmonar fueron normales.
El paciente se presentó para la evaluación actual a
pedido de sus dos nietos adolescentes, quienes querían saber sobre cualquier factor
de riesgo cardíaco familiar que pudiera influir en su participación en
deportes. Una revisión de los sistemas se destacó por palpitaciones
intermitentes, ojos secos crónicos, audición reducida, tinnitus en ambos oídos
(mayor en el oído derecho que en el oído izquierdo), piernas inquietas y
estreñimiento. El paciente no refirió dolor de cabeza, parestesia, síntomas de
accidente cerebrovascular, diarrea, cambios en la piel o intolerancia al calor
o al frío. Su historial médico incluía cardiomiopatía, enfermedad arterial
coronaria, bloqueo auriculoventricular y fibrilación auricular, dislipidemia,
hipertensión arterial, insuficiencia renal progresiva, cataratas e hipertrofia
prostática con carcinoma in situ. Los medicamentos incluyeron furosemida, warfarina,
amiodarona, aspirina, atorvastatina, metoprolol, enalapril y tamsulosina, así
como fluticasona inhalada. La amiodarona había causado tiroiditis, que había
sido tratada con prednisona; no hubo otras reacciones adversas conocidas a la
medicación.
El paciente era un ejecutivo jubilado y vivía con su
cónyuge en Massachusetts. Su historial familiar se destacaba por cáncer
cerebral en su padre, enfermedad pulmonar obstructiva y cáncer de pulmón en su
madre, y cáncer de pulmón en su hermana, quien había muerto a causa de la
enfermedad. También había antecedentes de miocardiopatía hipertrófica en su
hermana y en una sobrina. Su familia era de origen mediterráneo; la información
sobre otros parientes era limitada. Su hijo y sus nietos estaban sanos y, según
los informes, su hijo había tenido resultados normales en un electrocardiograma
y ecocardiogramas. El paciente no bebía alcohol y nunca había consumido tabaco.
En el examen, la frecuencia cardíaca era de 79 latidos
por minuto y la presión arterial de 106/60 mm Hg. El peso fue de 89 kg y el
índice de masa corporal 26,6. Había un
tirón del ventrículo izquierdo. La auscultación reveló desdoblamiento del ruido
cardíaco S2, un soplo sistólico grado 3/6 que se auscultaba en todo el
precordio y no cambiaba durante la maniobra de Valsalva, y un soplo
holosistólico grado 2/6 en el ápex que irradiaba a la axila; no había sonido
cardíaco S3. Había trazas de edema en las piernas y estasis venosa crónica
leve. El resto del examen fue normal.
Los resultados de las pruebas de laboratorio fueron
notables por un nivel de creatinina de 1,5 mg por decilitro, un nivel de
nitrógeno ureico de 39 mg por decilitro ( rango de referencia, 8 a 25 mg por
decilitro), una razón normalizada internacional de 2,1 (rango de referencia,
0,9 a 1,1), y un nivel de péptido natriurético tipo B N-terminal de 2523 pg por
mililitro (rango de referencia, 0 a 1800), así como 1+ proteína en el análisis
de orina. Los niveles sanguíneos de hierro, hemoglobina glucosilada,
tirotropina y tiroxina libre eran normales y la prueba de anticuerpos
antinucleares fue negativa. Un electrocardiograma mostró fibrilación auricular
con estimulación ventricular.
Se obtuvo un ETT y se realizaron pruebas de
diagnóstico adicionales.
Diagnóstico diferencial
Este hombre de 78 años se presentó para evaluación de
engrosamiento crónico de la pared miocárdica ventricular, regurgitación mitral
y taquiarritmias ventriculares recurrentes. Al determinar el proceso subyacente
a su miocardiopatía, es importante considerar las pistas de los ecocardiogramas
en el contexto de la historia y los hallazgos del examen físico. Revisé e
interpreté las imágenes ecocardiográficas más recientes del paciente, que se
obtuvieron en el momento de la evaluación actual. Las imágenes destacaban por
un severo engrosamiento simétrico de la pared miocárdica del ventrículo
izquierdo, que medía 21 mm en telediástole ( Figura 2A y 2B). El grosor de la
pared del ventrículo izquierdo varía según el sexo y la superficie corporal,
pero suele ser de 11 mm o menos. 1 Hubo deterioro limítrofe de la función
sistólica global.

Figura 2. Ecocardiogramas.
Un ecocardiograma transtorácico (ETT) en la vista de
eje largo paraesternal (Panel A) muestra el ventrículo izquierdo con marcado engrosamiento
de la pared, engrosamiento de la válvula aórtica y mitral, trazas de derrame
pericárdico y agrandamiento de la aurícula izquierda; la porción proximal de la
aorta no está dilatada y no hay borramiento en la unión sinotubular. Un ETT en
el eje corto paraesternal al nivel de la válvula mitral (Panel B) muestra que
el engrosamiento de la pared del ventrículo izquierdo es simétrico Un
ecocardiograma Doppler de onda continua a través de la válvula aórtica (Panel
C) muestra un pico velocidad de 1,5 m por segundo (rango de referencia en el
laboratorio de la Mayo Clinic, 0,8 a 1,8), hallazgo que indica la ausencia de
estenosis clínicamente significativa. Un ETT en el eje largo paraesternal con
Doppler color (Panel D) no muestra aceleración del flujo en el tracto de salida
del ventrículo izquierdo y no hay evidencia de movimiento anterior sistólico de
la válvula mitral; hay regurgitación mitral moderada, que se dirige centralmente,
en lugar de posteriormente dirigido (como se ve típicamente con la
miocardiopatía hipertrófica). Un ecocardiograma Doppler de onda pulsada deL flujo
de entrada mitral (Panel E) muestra una velocidad máxima elevada durante la
diástole temprana (velocidad de onda E, 120 cm por segundo; rango de
referencia, 40 a 88), que se debe en parte a una regurgitación mitral
clínicamente significativa, así como a un tiempo de desaceleración marcadamente
abreviado, lo que sugiere de un ventrículo izquierdo rígido y predictivo de un
aumento de presión de llenado ventricular. Un ecocardiograma Doppler tisular
del anillo mitral lateral (Panel F) muestra una velocidad severamente
disminuida durante la diástole temprana (velocidad e′, 5 cm por segundo; valor
de referencia, >9). La relación notablemente aumentada entre la velocidad de
la onda E y la velocidad e′ (relación E:e′, 24; valor de referencia, <10),
además del agrandamiento biauricular y una presión sistólica estimada del
ventrículo derecho de 56 mm Hg (no se muestra), es consistente con disfunción
diastólica restrictiva del ventrículo izquierdo
ENGROSAMIENTO DE LA PARED DEL VENTRÍCULO IZQUIERDO
La causa más frecuente de engrosamiento de la pared
del ventrículo izquierdo es la cardiopatía hipertensiva, que se desarrolla en
el contexto de una hipertensión de larga evolución mal controlada. Los
pacientes con hipertensión pueden tener hipertrofia concéntrica, hipertrofia
excéntrica o remodelación concéntrica en el ventrículo izquierdo. Estos tipos
de remodelado se diferencian según el índice de masa ventricular izquierda y el
grosor parietal relativo; la presencia de obesidad concurrente, cardiopatía
valvular o isquémica y factores genéticos pueden influir en el tipo de
remodelación que se produce. 2,3El engrosamiento de la pared del ventrículo
izquierdo asociado con hipertensión es reversible. Este paciente había recibido
tratamiento para la hipertensión y su presión arterial era normal en el momento
de la evaluación actual. Además, no estaba presente otra característica de la
cardiopatía hipertensiva, el borramiento de la unión sinotubular ( Figura 2A ).
El engrosamiento de la pared del ventrículo izquierdo
puede ser consecuencia del entrenamiento deportivo, dependiendo del tipo de
ejercicio. Los atletas pueden tener aumentos en el diámetro, el grosor de la
pared y la masa del ventrículo izquierdo que se asocian con una función
sistólica y diastólica normal y con bradicardia sinusal. 4 Un grosor de la
pared del ventrículo izquierdo de 13 mm o más es poco común en los atletas. El
grosor de la pared generalmente disminuye con el desentrenamiento, aunque el
corazón permanece agrandado en algunos atletas. Este paciente tenía
antecedentes extensos de engrosamiento de la pared del ventrículo izquierdo,
que progresó mucho después de que dejó de practicar deportes; este factor
sugiere que un proceso subyacente diferente estaba causando el engrosamiento de
la pared del ventrículo izquierdo.
La hipertrofia ventricular izquierda puede ser causada
por estenosis de la válvula aórtica, estenosis aórtica subvalvular, estenosis
aórtica supravalvular o coartación aórtica, pero estas anomalías se habrían
detectado en la ecocardiografía. El paciente tenía engrosamiento de la válvula
aórtica de tres valvas, pero la ecocardiografía Doppler no reveló una
obstrucción de la válvula aórtica clínicamente significativa ( Figura 2C ).
Las condiciones que se han considerado hasta ahora no
explicarían el engrosamiento simultáneo de la pared miocárdica del ventrículo
derecho, que se visualiza mejor en la vista subcostal. Por lo tanto, las
condiciones asociadas con el engrosamiento de la pared biventricular deben
considerarse en el diagnóstico diferencial de este paciente.
ENGROSAMIENTO DE LA PARED BIVENTRICULAR
Los antecedentes de miocardiopatía hipertrófica en la
hermana y la sobrina de la paciente sugieren un trastorno genético familiar. La
miocardiopatía hipertrófica convencional es una enfermedad autosómica dominante
causada por mutaciones en las proteínas sarcoméricas. Tiene una prevalencia
estimada de 1 en 500 personas, pero la prevalencia puede ser mayor que la
estimada debido a la enfermedad no diagnosticada. 5 La miocardiopatía
hipertrófica se asocia más comúnmente con la hipertrofia ventricular que afecta
solo al ventrículo izquierdo. Sin embargo, existe una heterogeneidad fenotípica
considerable y puede ocurrir hipertrofia del ventrículo derecho, lo que puede
dificultar el establecimiento del diagnóstico. La obstrucción del tracto de
salida del ventrículo izquierdo está presente o se desarrolla con el tiempo en
el 70% de los pacientes con miocardiopatía hipertrófica. 5La hipertrofia o el
desplazamiento de los músculos papilares, la inserción anómala de los músculos
papilares y el alargamiento de las valvas de la válvula mitral con movimiento
anterior sistólico e insuficiencia mitral dirigida hacia atrás son
características ecocardiográficas frecuentes. Aunque esta paciente tenía
hipertrofia del músculo papilar, tenía engrosamiento de la válvula mitral e
insuficiencia mitral dirigida centralmente ( Figura 2D ).
El edema miocárdico puede contribuir al engrosamiento
de la pared ventricular. Puede ocurrir en pacientes que se encuentran en un
estado edematoso sistémico por insuficiencia renal o insuficiencia cardiaca o
en pacientes con miocarditis aguda. Sin embargo, el grado de engrosamiento de
la pared miocárdica asociado con edema o miocarditis es más leve que el
observado en este paciente. Su presentación clínica no era compatible con un
estado edematoso o con una enfermedad aguda.
ENFERMEDADES SISTÉMICAS ASOCIADAS AL ENGROSAMIENTO DE
LA PARED VENTRICULAR
¿Tiene este paciente un trastorno multiorgánico
sistémico asociado con engrosamiento de la pared biventricular? Tiene
antecedentes de hipohidrosis, audición reducida, ojos secos crónicos,
estreñimiento e insuficiencia renal, todos los cuales pueden ser características
de una enfermedad sistémica.
El engrosamiento de la pared biventricular se puede
asociar con amiloidosis cardíaca, que ocurre como parte de una enfermedad
sistémica, ya sea amiloidosis de cadena ligera (AL) o amiloidosis por
transtiretina. Las características de la amiloidosis cardíaca pueden incluir
edema, ascitis y disnea debido a una miocardiopatía restrictiva; fatiga por
bajo gasto cardíaco; y bradiarritmias y bloqueo cardiaco. La insuficiencia
cardíaca generalmente progresa rápidamente, especialmente en pacientes con
amiloidosis AL cardíaca. La afectación de los tejidos blandos a menudo es
evidente en el examen físico con amiloidosis AL o amiloidosis por
transtiretina. 8
La amiloidosis AL está causada por una discrasia de
células plasmáticas subyacente. La amiloidosis por transtiretina se desarrolla
cuando una proteína producida por el hígado se vuelve inestable, lo que hace
que se acumulen depósitos de fibrillas de amiloide en el corazón, los nervios y
otros órganos. Existe una forma autosómica dominante de amiloidosis cardíaca
por transtiretina que es más frecuente en personas de origen afrocaribeño, así
como una forma esporádica que puede desarrollarse en los ancianos. 9
Una característica llamativa de la amiloidosis
cardíaca es la discrepancia entre el grosor de la pared observado en la
ecocardiografía y el voltaje del QRS observado en la electrocardiografía; el
voltaje del QRS está típicamente reducido, aunque puede haber un patrón de
seudoinfarto. La característica de la enfermedad de este paciente que hace que
la amiloidosis cardíaca sea muy poco probable es su naturaleza indolente. La
mediana de supervivencia asociada con la amiloidosis cardíaca es de menos de 4
años entre los pacientes no tratados y es aún más corta entre los que tienen amiloidosis
AL. 10 Cabe señalar que otras enfermedades sistémicas, como la hemocromatosis y
la sarcoidosis, se asocian con la miocardiopatía dilatada, más que con la
miocardiopatía hipertrófica.
TRASTORNOS GENÉTICOS ASOCIADOS CON EL ENGROSAMIENTO DE
LA PARED VENTRICULAR
Hay varios trastornos sistémicos hereditarios que se
asocian con hipertrofia miocárdica pero que también causan otras dismorfias no
cardíacas y se manifiestan en etapas tempranas de la vida. Estos incluyen el
síndrome de Noonan y otros trastornos resultantes de mutaciones genéticas en la
vía de señalización RAS-MAPK (conocidas como RASopatías). 11 Las miopatías
mitocondriales se pueden asociar con hipertrofia ventricular izquierda, pero
los pacientes típicamente se presentan a una edad más temprana con debilidad
muscular proximal, miopatía ocular y bloqueo cardíaco; la crisis mitocondrial
puede ser precipitada por el estrés. 12,13 La ataxia de Friedreich, que es una
enfermedad autosómica recesiva resultante de un defecto en FXN (el gen que codifica
la proteína mitocondrial frataxina), puede causar hipertrofia ventricular
izquierda, 14pero los pacientes suelen tener ataxia por daño del sistema
nervioso. La enfermedad por almacenamiento de glucógeno tipo III, que resulta
de una deficiencia en la enzima desramificadora de glucógeno, puede causar
hipertrofia del ventrículo izquierdo, pero los pacientes suelen presentar en la
infancia hipoglucemia, baja estatura y hepatomegalia. 15
Los trastornos de almacenamiento lisosomal que se
asocian con el engrosamiento de la pared del miocardio incluyen la enfermedad
de Danon, la enfermedad de Pompe, las mucopolisacaridosis y la enfermedad de
Fabry. La enfermedad de Danon (o miocardiopatía de Danon) es una enfermedad
dominante ligada al cromosoma X que resulta de defectos genéticos en LAMP2 (el
gen que codifica la proteína 2 de la membrana asociada a los lisosomas) que
provoca un engrosamiento de la pared miocárdica de moderado a grave, miopatía
esquelética y discapacidad intelectual. dieciséisLa enfermedad de Pompe es un
trastorno autosómico recesivo causado por una deficiencia de α-glucosidasa
ácida, que conduce a la acumulación intralisosomal de glucógeno y a un aumento
de la masa miocárdica. Tanto en niños como en adultos con este trastorno, la
debilidad de los músculos esqueléticos y la afectación de los músculos
respiratorios son características destacadas. Las mucopolisacaridosis están
causadas por defectos de la degradación intralisosomal de glucosaminoglucanos.
Además de la miocardiopatía y el engrosamiento de las válvulas mitral y
aórtica, la calcificación del anillo mitral y el engrosamiento de la piel y las
características faciales son hallazgos destacados y no estaban presentes en
este paciente.
ENFERMEDAD DE FABRY
La enfermedad de Fabry es un trastorno hereditario
ligado al cromosoma X causado por una deficiencia de α-galactosidasa A
lisosomal, que conduce a la acumulación de glucoesfingolípidos ( Figura 3 ).
Los antecedentes de miocardiopatía hipertrófica en la hermana y la sobrina del
paciente, pero no en su hijo, podrían ser compatibles con un trastorno
hereditario ligado al cromosoma X. La enfermedad de Fabry es rara y, a menudo,
hay un retraso entre el inicio de los síntomas y el diagnóstico, con un retraso
medio de 14 años en los hombres. 17 Una manifestación clínica común, y una
pista potencialmente importante en este caso, es la hipohidrosis. Otra
manifestación común es la hipertensión sistémica, que a menudo se debe a
insuficiencia renal coexistente. La acumulación de globotriaosilceramida en
cardiomiocitos, células endoteliales y células de músculo liso conduce a
isquemia miocárdica, anomalías valvulares y engrosamiento de la pared
miocárdica. Se puede desarrollar disfunción diastólica ( Figura 2E y 2F ) e
insuficiencia cardíaca. Las arritmias y la enfermedad del sistema de conducción
ocurren comúnmente en pacientes con la enfermedad de Fabry.En este paciente, el
curso prolongado y gradualmente progresivo del engrosamiento de la pared del
miocardio, las arritmias asociadas y la enfermedad del sistema de conducción,
el engrosamiento de la válvula, la afectación familiar (que aparentemente es
más grave en el miembro masculino afectado de la familia) y la enfermedad renal
concurrente y hipohidrosis hacen que la enfermedad de Fabry sea el diagnóstico
más probable.

Figura 3. Manifestaciones clínicas longitudinales de
la enfermedad de Fabry en este paciente.
Se muestran las manifestaciones de la enfermedad de
Fabry que estaban presentes cuando el paciente tenía aproximadamente 25 años de
edad, así como los que estuvieron presentes al momento de su actual evaluación
IMPRESIÓN CLÍNICA
Cuando evaluamos a este paciente, inicialmente
consideramos que la miocardiopatía hipertrófica era el diagnóstico más
probable, dado el grado de engrosamiento de la pared ventricular, las
taquiarritmias ventriculares, la fibrilación auricular y los antecedentes
familiares informados. Las características que serían atípicas de la
miocardiopatía hipertrófica sarcomérica convencional estaban presentes,
incluida la naturaleza concéntrica y biventricular del engrosamiento de la
pared y, en cierta medida, el bloqueo cardíaco.
Los factores genéticos que causan el engrosamiento de
la pared del ventrículo izquierdo pueden ser monogénicos (que involucran una
variación genética en un solo gen) o poligénicos (influidos por variantes que
involucran múltiples genes). Las pruebas genéticas para el engrosamiento de la
pared del ventrículo izquierdo generalmente están indicadas cuando existe un
alto índice de sospecha de miocardiopatía hipertrófica. El índice de sospecha
se puede estimar mediante la obtención de una historia familiar detallada que
incluya al menos tres generaciones. En este caso, la hermana y la sobrina de la
paciente habían recibido el diagnóstico de miocardiopatía hipertrófica. Aunque
los detalles son limitados, esta historia aumenta el índice de sospecha de un
trastorno monogénico, quizás un trastorno autosómico dominante, como la
miocardiopatía hipertrófica sarcomérica convencional.Sin embargo, la ausencia
de transmisión informada entre miembros masculinos de la familia plantea la
posibilidad de una afección ligada al cromosoma X, como la enfermedad de Fabry.
Una advertencia importante es que una evaluación familiar está incompleta sin
un fenotipo clínico detallado. Finalmente, este paciente fue derivado a un
asesor genético para evaluación, asesoramiento, pruebas genéticas, divulgación
de resultados y asesoramiento posterior a la prueba.
DIAGNOSTICO CLINICO
MIOCARDIOPATÍA HIPERTRÓFICA MUY PROBABLEMENTE DEBIDO A
LA ENFERMEDAD DE FABRY.
PRUEBAS DE DIAGNÓSTICO
La prueba de diagnóstico en este caso fue la
secuenciación de 26 genes que se sabe que causan la miocardiopatía
hipertrófica. Las pruebas genéticas revelaron una mutación sin sentido en GLA
(el gen que codifica la α-galactosidasa A): c.901C→G (p.Arg301Gly) en el exón
6. Este resultado fue consistente con un diagnóstico de enfermedad de Fabry. El
diagnóstico se confirmó aún más con la prueba de α-galactosidasa A en plasma,
que no reveló actividad residual (0,0001 U por litro; rango de referencia,
0,074 a 0,457).
La enfermedad de Fabry resulta de una deficiencia en
la actividad de la enzima glicohidrolasa α-galactosidasa A. La anormalidad
primaria implica la acumulación de globotriaosilceramida en una variedad de
tipos de células, que comienza en el útero en los casos más graves. Esta
acumulación de globotriaosilceramida progresa con el tiempo y conduce al daño
de los órganos diana. Los órganos que se ven afectados con mayor frecuencia
incluyen el corazón (engrosamiento de la pared del ventrículo izquierdo,
arritmias y fibrosis), los riñones (glomeruloesclerosis y proteinuria) y el
sistema nervioso central y periférico (cambios en la sustancia blanca,
accidentes cerebrovasculares y neuropatías). Si no se trata, la acumulación de
globotriaosilceramida puede provocar una muerte prematura. 18 En los casos
clásicos de la enfermedad de Fabry, el inicio de los síntomas ocurre en la
niñez y las complicaciones surgen en la edad adulta ( Figura 3 ). 19
DIAGNÓSTICO GENÉTICO
ENFERMEDAD DE FABRY.
ECOCARDIOGRAFÍA EN LA ENFERMEDAD DE FABRY
Las imágenes cardíacas son una herramienta importante
en el diagnóstico y manejo de las manifestaciones cardíacas de la enfermedad de
Fabry. La ecocardiografía es fundamental porque los hallazgos pueden impulsar
la evaluación de la actividad de la α-galactosidasa A y las pruebas genéticas
en personas que no han recibido previamente un diagnóstico de enfermedad de
Fabry. Después del diagnóstico, la ecocardiografía es la opción lógica de
imagen para monitorear la progresión de la enfermedad y la respuesta al
tratamiento.
Las principales características ecocardiográficas
bidimensionales observadas en este paciente con enfermedad de Fabry ( Tabla 1 )
incluyeron un engrosamiento simétrico de la pared del ventrículo izquierdo
clínicamente significativo ( Figuras 2A y 2B ). También se presentó
engrosamiento de la pared del ventrículo derecho. La función diastólica estaba
gravemente afectada ( Figura 2E y 2F). Aunque la función sistólica
biventricular evaluada en imágenes bidimensionales o tridimensionales a menudo
se conserva hasta etapas posteriores, las anomalías diastólicas y el
agrandamiento biauricular son comunes. También es común el engrosamiento
valvular con regurgitación. También se ha descrito dilatación del seno aórtico
y de la aorta ascendente en pacientes con enfermedad de Fabry. Ninguno de estos
hallazgos ecocardiográficos, solos o en combinación, puede confirmar o
descartar el diagnóstico de la enfermedad de Fabry. El signo binario, que
previamente se asoció con la enfermedad de Fabry, se refiere a la presencia de
un endocardio brillante con una capa subendocárdica hiporreflectante adyacente,
de modo que puede distinguirse de la pared media del miocardio al final de la
diástole. La evidencia más reciente ha demostrado que este hallazgo no es
sensible ni específico para la enfermedad de Fabry.
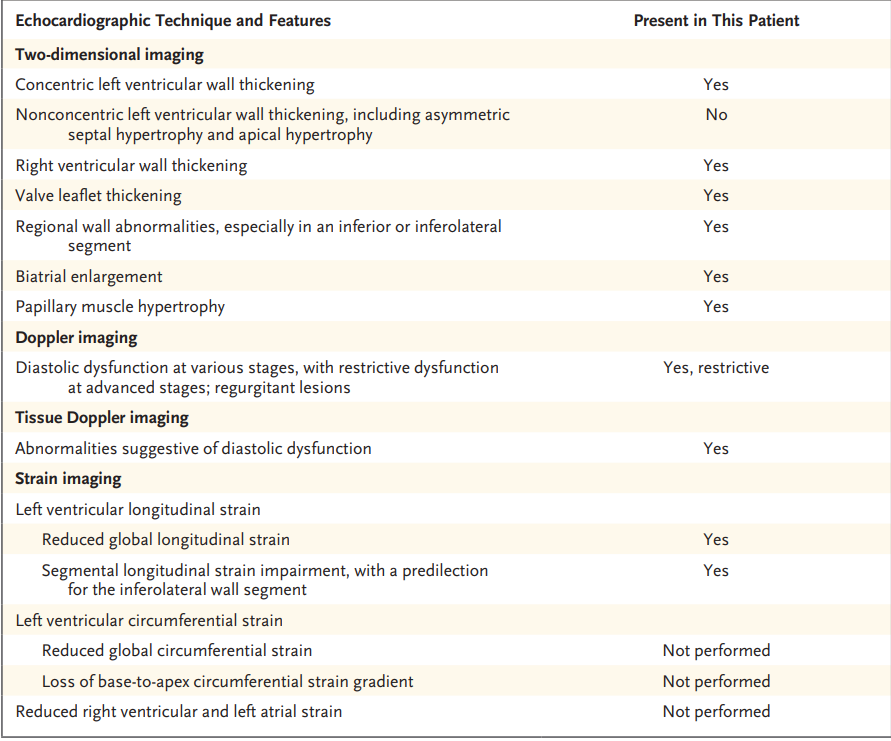
Tabla 1. Características ecocardiográficas clave de la
enfermedad de Fabry.
Las imágenes de tensión tienen un papel importante en
la reducción del diagnóstico diferencial de la miocardiopatía. Cuando se
sospecha la enfermedad de Fabry, se pueden realizar imágenes de tensión para
evaluar dos patrones que sugieran la enfermedad. El primer patrón es el
deterioro de la tensión longitudinal segmentaria que afecta a la pared
inferolateral basal. 20,21 Esto puede ir acompañado de una tensión longitudinal
global reducida. Aunque cualquier segmento puede verse afectado en pacientes
con enfermedad de Fabry, existe una predilección por el segmento de la pared
inferolateral. Un análisis de tensión retrospectivo de un ecocardiograma
obtenido de este paciente, que se realizó en preparación para esta conferencia,
mostró una marcada reducción de la tensión longitudinal global, a un nivel
promedio de −5,6% (valor de referencia, menos de −18) 22 .siendo los segmentos
inferolateral y septal los más severamente afectados ( Video 6 ). Este hallazgo
destaca la presencia de disfunción contráctil miocárdica severa, a pesar de la
fracción de eyección del ventrículo izquierdo normal baja evaluada en la
ecocardiografía bidimensional. El segundo patrón a buscar en las imágenes de
deformación en pacientes con sospecha de enfermedad de Fabry es una pérdida del
gradiente de deformación circunferencial de la base al ápice. 23
IMÁGENES CARDÍACAS ADICIONALES EN LA ENFERMEDAD DE
FABRY
Aunque a este paciente no se le realizó una resonancia
magnética cardíaca, esta es una herramienta importante para la evaluación del
compromiso cardíaco en pacientes con la enfermedad de Fabry, así como para la
evaluación del engrosamiento indiferenciado de la pared ventricular. 24,25 Al
principio de la enfermedad de Fabry, los pacientes suelen tener un
engrosamiento simétrico de la pared del ventrículo izquierdo con una fracción
de eyección del ventrículo izquierdo preservada. 26 Más adelante en el proceso
de la enfermedad, los pacientes pueden tener adelgazamiento del segmento de la
pared inferolateral basal con la correspondiente hipocinesia o anomalías más
difusas del movimiento de la pared con una fracción de eyección reducida. 27
El mapeo T1 nativo y las imágenes tardías después de
la administración de gadolinio permiten una mayor caracterización del tejido y
ayudan en el diagnóstico. El realce tardío de gadolinio en el miocardio medio
generalmente se encuentra dentro de los segmentos de la pared inferolateral basal
y media y puede estar presente hasta en el 20% de las pacientes con enfermedad
de Fabry que no tienen engrosamiento de la pared del ventrículo izquierdo.
28Los tiempos T1 nativos bajos pueden estar presentes antes del desarrollo del
engrosamiento de la pared del ventrículo izquierdo, lo que lleva a la
identificación de compromiso cardíaco temprano en pacientes con enfermedad de
Fabry. En pacientes con engrosamiento indiferenciado de la pared del ventrículo
izquierdo, los tiempos T1 nativos bajos pueden sugerir un diagnóstico de
enfermedad de Fabry, porque este hallazgo está presente en aproximadamente el
90 % de los pacientes con enfermedad de Fabry y engrosamiento de la pared del
ventrículo izquierdo. 29
DISCUSIÓN DEL MANEJO
La progresión de la enfermedad de Fabry sigue un curso
que es único para cada paciente afectado. Un enfoque individualizado del manejo
comienza con una evaluación y un seguimiento completos de cada compartimiento
de órganos. 30Se encontró que este paciente tenía pérdida de audición,
tinnitus, cataratas, cambios en la sustancia blanca en las imágenes y un nivel
elevado de creatinina con proteinuria leve, todos los cuales son hallazgos
compatibles con la enfermedad de Fabry. El manejo incluyó intervenciones
basadas en los síntomas para su enfermedad cardíaca y renal, evaluaciones
anuales de órganos terminales y atención de apoyo brindada por un equipo
multidisciplinario con experiencia en el tratamiento de la enfermedad de Fabry.
Se ofreció una terapia específica para la enfermedad de Fabry, incluidas
opciones para buscar terapia de reemplazo de enzimas o terapia de chaperonas.
Dada la etapa avanzada del engrosamiento de la pared del ventrículo izquierdo
en este paciente, era poco probable que dicha terapia revirtiera el daño cardíaco
y detuviera la progresión de la enfermedad. Aunque este tratamiento pudo haber
protegido otros órganos con riesgo de afectación de la enfermedad, el paciente
rechazó el tratamiento.31 El paciente estaba tomando amiodarona, que es un
inhibidor competitivo de la fosfolipasa A 2 en el lisosoma y puede exacerbar
los síntomas de la enfermedad de Fabry. 32 Se discutió la interrupción de la
terapia con amiodarona, pero el beneficio de su uso para controlar las
arritmias superó los posibles efectos negativos aditivos en la progresión de la
enfermedad de Fabry.
SEGUIMIENTO
Después de 3 años de seguimiento, los resultados de la
ecocardiografía no cambiaron. El paciente había sufrido una hospitalización por
insuficiencia cardíaca. Tenía eventos de taquicardia ventricular no sostenida
en curso, por lo que se ajustaron los regímenes de metoprolol y amiodarona.
Pudo viajar y celebrar un hito importante en su vida con su familia.
DIAGNOSTICO FINAL
ENFERMEDAD DE FABRY.
Traducción de
A 78-Year-Old Man with Marked Ventricular Wall
Thickening
Patricia A. Pellikka, M.D., David M. Dudzinski, M.D.,
Steven A. Lubitz, M.D., M.P.H., Teresa S.M. Tsang, M.D., Albree Tower-Rader,
M.D., and Amel Karaa, M.D.
https://www.nejm.org/doi/full/10.1056/NEJMcpc2201230
References
1. Lang RM, Badano LP, Mor-Avi V, et al.
Recommendations for cardiac chamber
quantification by echocardiography in
adults: an update from the American So[1]ciety
of Echocardiography and the Euro[1]pean
Association of Cardiovascular Imag[1]ing.
J Am Soc Echocardiogr 2015;28(1):
1-39.e14.
2. Ganau A, Devereux RB, Roman MJ,
et al. Patterns of left ventricular hypertro[1]phy
and geometric remodeling in essen[1]tial
hypertension. J Am Coll Cardiol 1992;
19:1550-8.
3. Drazner MH. The progression of hyper[1]tensive
heart disease. Circulation 2011;
123:327-34.
4. Fagard R. Athlete’s heart. Heart 2003;
89:1455-61.
5. Geske JB, Ommen SR, Gersh BJ. Hyper[1]trophic
cardiomyopathy: clinical update.
JACC Heart Fail 2018;6:364-75.
6. Amann K, Rychlík I, Miltenberger[1]Milteny
G, Ritz E. Left ventricular hyper[1]trophy
in renal failure. Kidney Int Suppl
1998;68:S78-S85.
7. Hiramitsu S, Morimoto S, Kato S,
et al. Transient ventricular wall thicken[1]ing
in acute myocarditis: a serial echocar[1]diographic
and histopathologic study. Jpn
Circ J 2001;65:863-6.
8. Sanchorawala V. Light-chain (AL)
amyloidosis: diagnosis and treatment.
Clin J Am Soc Nephrol 2006;1:1331-41.
9. Dungu JN, Anderson LJ, Whelan CJ,
Hawkins PN. Cardiac transthyretin amy[1]loidosis.
Heart 2012;98:1546-54.
10. Kittleson MM, Maurer MS, Ambardekar
AV, et al. Cardiac amyloidosis: evolving
diagnosis and management: a scientific
statement from the American Heart Asso[1]ciation.
Circulation 2020;142(1):e7-e22.
11. Calcagni G, Adorisio R, Martinelli S,
et al. Clinical presentation and natural
history of hypertrophic cardiomyopathy
in RASopathies. Heart Fail Clin 2018;14:
225-35.
12. Anan R, Nakagawa M, Miyata M, et al.
Cardiac involvement in mitochondrial dis[1]eases:
a study on 17 patients with docu[1]mented
mitochondrial DNA defects. Cir[1]culation
1995;91:955-61.
13. Meyers DE, Basha HI, Koenig MK.
Mitochondrial cardiomyopathy: patho[1]physiology,
diagnosis, and management.
Tex Heart Inst J 2013;40:385-94.
14. Weidemann F, Rummey C, Bijnens B,
et al. The heart in Friedreich ataxia: defi[1]nition
of cardiomyopathy, disease severity,
and correlation with neurological symp[1]toms.
Circulation 2012;125:1626-34.
15. Vertilus SM, Austin SL, Foster KS, et al.
Echocardiographic manifestations of Gly[1]cogen
Storage Disease III: increase in wall
thickness and left ventricular mass over
time. Genet Med 2010;12:413-23.
16. D’souza RS, Levandowski C, Slavov D,
et al. Danon disease: clinical features,
evaluation, and management. Circ Heart
Fail 2014;7:843-9.
17. Mehta A, Ricci R, Widmer U, et al.
Fabry disease defined: baseline clinical
manifestations of 366 patients in the
Fabry Outcome Survey. Eur J Clin Invest
2004;34:236-42.
18. Schiffmann R. Fabry disease. Phar[1]macol
Ther 2009;122:65-77.
19. Mehta A, Hughes DA. Fabry disease.
In: Adam MP, Ardinger HH, Pagon RA,
Wallace SE, eds. GeneReviews. Seattle:
University of Washington, 2002 (http://
www.ncbi.nlm.nih.gov/books/NBK1292/).
20. Yeung DF, Sirrs S, Tsang MYC, et al.
Echocardiographic assessment of patients
with Fabry disease. J Am Soc Echocar[1]diogr
2018;31(6):639-649.e2.
21. Zada M, Lo Q, Boyd AC, et al. Basal
segmental longitudinal strain: a marker
of subclinical myocardial involvement in
Anderson-Fabry disease. J Am Soc Echo[1]cardiogr
2021;34(4):405-413.e2.
22. Yingchoncharoen T, Agarwal S,
Popović ZB, Marwick TH. Normal ranges
of left ventricular strain: a meta-analysis.
J Am Soc Echocardiogr 2013;26:185-91.
23. Labombarda F, Saloux E, Milesi G,
Bienvenu B. Loss of base-to-apex circum[1]ferential
strain gradient: a specific pat[1]tern
of Fabry cardiomyopathy? Echocar[1]diography
2017;34:504-10.
24. Ortiz A, Germain DP, Desnick RJ, et al.
Fabry disease revisited: management and
treatment recommendations for adult pa[1]tients.
Mol Genet Metab 2018;123:416-27.
25. Ommen SR, Mital S, Burke MA, et al.
2020 AHA/ACC guideline for the diagno[1]sis
and treatment of patients with hyper[1]trophic
cardiomyopathy: a report of the
American College of Cardiology/Ameri[1]can
Heart Association Joint Committee
on Clinical Practice Guidelines. J Am Coll
Cardiol 2020;76(25):e159-e240.
26. Linhart A, Elliott PM. The heart in
Anderson-Fabry disease and other lyso[1]somal
storage disorders. Heart 2007;93:
528-35.
27. Kawano M, Takenaka T, Otsuji Y, et al.
Significance of asymmetric basal poste[1]rior
wall thinning in patients with car[1]diac
Fabry’s disease. Am J Cardiol 2007;
99:261-3.
28. Niemann M, Herrmann S, Hu K, et al.
Differences in Fabry cardiomyopathy be[1]tween
female and male patients: conse[1]quences
for diagnostic assessment. JACC
Cardiovasc Imaging 2011;4:592-601.
29. Pica S, Sado DM, Maestrini V, et al.
Reproducibility of native myocardial T1
mapping in the assessment of Fabry dis[1]ease
and its role in early detection of car[1]diac
involvement by cardiovascular mag[1]netic
resonance. J Cardiovasc Magn Reson
2014;16:99.
30. Eng CM, Germain DP, Banikazemi M,
et al. Fabry disease: guidelines for the evalu[1]ation
and management of multi-organ
system involvement. Genet Med 2006;8:
539-48.
31. Schiffmann R. Fabry disease. Handb
Clin Neurol 2015;132:231-48.
32. Bracamonte ER, Kowalewska J, Starr
J, Gitomer J, Alpers CE. Iatrogenic phos[1]pholipidosis
mimicking Fabry disease.
Am J Kidney Dis 2006;48:844-50.