Un hombre de 59 años fue evaluado en la clínica de reumatología de este hospital debido a dolor de cabeza y disfunción neurológica progresiva.
Diez meses antes de esta evaluación, desarrolló fiebre
diaria con temperatura de hasta 38,9°C, escalofríos, mialgias, sudoración
nocturna profusa, presión y dolor en ambas mejillas y oídos. El paciente fue
evaluado por su médico de atención primaria. Informó rinorrea y fatiga, pero no
dolor de cabeza, faringitis o disnea, y no tenía contactos enfermos. En el
examen, la temperatura era de 37,8°C, la frecuencia cardíaca de 100 latidos por
minuto, la presión arterial de 112/64 mm Hg y la saturación de oxígeno del 98%
mientras respiraba aire ambiente. Tenía mal aspecto y los senos maxilares
estaban sensibles a la palpación; El resto del examen era normal. Los niveles
sanguíneos de electrolitos, calcio, proteína total, albúmina y globulina eran
normales, al igual que los resultados de las pruebas de función renal. El recuento
de plaquetas fue de 463.000 por microlitro (rango de referencia, 140.000 a 400,
000); el resto del conteo sanguíneo completo y el conteo diferencial fueron
normales. Se prescribió amoxicilina-clavulanato.
Durante las siguientes 2 semanas, continuaron la
fiebre, los sudores nocturnos y el dolor facial y de los senos paranasales.
Desarrolló dolor de cabeza difuso, junto con dolor en la mandíbula,
sensibilidad en el cuero cabelludo y rigidez en la cadera y los hombros. El
paciente fue evaluado nuevamente por su médico de atención primaria. Refirió
pérdida de peso de 2 kg, visión doble episódica y dolor en la cadera que le
provocaba dificultad para ponerse de pie y subir escaleras. El examen físico no
se modificó. El análisis de orina fue normal. Una prueba de anticuerpos
heterófilos y un ensayo de liberación de interferón-γ para Mycobacterium
tuberculosis fueron negativos, al igual que las pruebas de detección del virus
de la inmunodeficiencia humana y la enfermedad de Lyme. La radiografía de tórax
y la tomografía computarizada de senos paranasales fueron normales. El paciente
fue derivado a un reumatólogo en otro hospital.
En la evaluación, el paciente refirió fiebre
persistente y cefalea frontal, temporal y maxilar. Había pulsos palpables de la
arteria temporal sin sensibilidad; el resto de la exploración fue normal,
incluida la ausencia de soplos a la auscultación de grandes vasos. La velocidad
de sedimentación de eritrocitos fue de 108 mm por hora (rango de referencia, 0
a 20), el nivel en sangre de proteína C reactiva mayor a 100 mg por litro
(valor de referencia, <8,0), el nivel en sangre de factor reumatoideo 24 UI
por mililitro (valor de referencia, <14), y el nivel en sangre de aldolasa
8,4 UI por mililitro (valor de referencia, <8,1). Se realizó biopsia de
arteria temporal derecha. Según los informes, el examen histológico de la
muestra de biopsia no mostró granulomas.
Se realizó el diagnóstico de arteritis de células
gigantes y se inició tratamiento con prednisona. A los pocos días del inicio
del tratamiento, el dolor de cabeza, la fiebre, el dolor de mandíbula y la
rigidez de las articulaciones disminuyeron. Al mes siguiente, un intento de
reducir la dosis de prednisona resultó en un dolor de cabeza severo recurrente
con sensibilidad en el cuero cabelludo, dolor en la mandíbula y rigidez en la
cadera. La velocidad de sedimentación globular fue de 55 mm por hora y el nivel
de proteína C reactiva de 84 mg por litro. Se inició tratamiento con
administración subcutánea semanal de tocilizumab.
Durante las siguientes 6 semanas, se hizo otro intento
de disminuir la dosis de prednisona. Después de 1 semana de tratamiento con una
dosis más baja de prednisona y 5 meses después del desarrollo de los síntomas
iniciales (5 meses antes de la evaluación actual), el paciente informó dolor de
cabeza que describió como “el peor dolor de cabeza de mi vida”, con dolor de
mandíbula asociado y diplopía. El dolor era peor en las áreas frontal y maxilar
derechas, pero estaba afectado todo el lado derecho de la cara, incluido el
cuero cabelludo y el cuello. El paciente regresó al otro hospital para su
evaluación. Se encontró que tenía parálisis de los nervios craneales tercero y
sexto en el lado derecho. El paciente ingresó en el servicio de neurología del
otro hospital. La prueba de anticuerpos anticitoplasma de neutrófilos (ANCA)
reveló un nivel de anticuerpos mieloperoxidasa ligeramente elevado, a 1,9 U
(valor de referencia, < 1.0), pero no se detectaron anticuerpos contra la
proteinasa 3. Los niveles en sangre de las subclases de IgG eran normales. Se
obtuvieron estudios de imagen.
La resonancia magnética (RM) de la cabeza ( Figura 1A
y 1B ), realizada después de la administración de contraste intravenoso, reveló
una expansión anormal y realce del seno cavernoso derecho que se extendía hacia
la fisura orbitaria superior derecha y la fosa pterigopalatina derecha, así
como engrosamiento paquimeníngeo anormal y realce de la fosa craneal media
derecha adyacente. Resonancia magnética de la columna cervical ( Figura 1C y
1D) mostró engrosamiento y realce paquimeníngeo similar a una masa en el canal
espinal cervical superior en C2-C4 con predominio anterior, así como más
circunferencialmente en la unión cervicotorácica (C6-T3) con predominio
posterior, con estrechamiento del canal resultante y compresión leve de la
médula en C7 –T2. No había edema de la médula espinal.

Figura 1. Resonancia magnética de cabeza y columna
obtenida 5 meses antes de la evaluación actual.
Las mágenes axiales y coronales potenciadas en T1 con
contraste de la cabeza (Paneles A y B, respectivamente) muestran realce del
seno cavernoso derecho que se extiende hacia la fisura orbitaria superior
derecha y el tercio medio derecho adyacente de la fosa craneal (flechas).
Imágenes sagitales y axiales potenciadas en T1 con contraste de la columna
cervical con supresión grasa (Paneles C y D, respectivamente) muestran realce
paquimeníngeo anormal y engrosamiento anterior en el canal espinal cervical
superior en C2-C4, así como más circunferencialmente con un predominio posterior
en la unión cervicotorácica, con estenosis del canal variable resultante y
compresión leve de la médula (flechas).
Se realizó una punción lumbar; no se registró una
presión de apertura. El nivel de glucosa en líquido cefalorraquídeo (LCR) fue de
65 mg por decilitro ( rango de referencia, 50 a 80 mg por decilitro), y el
nivel de proteína fue de 126 mg por decilitro (rango de referencia , 15 a 45),
con 2 glóbulos rojos por microlitro y 8 glóbulos blancos por microlitro (de los
cuales el 79% eran linfocitos). La tinción de Gram y los cultivos de LCR fueron
negativos. Las pruebas de proteína básica de mielina del LCR, enzima
convertidora de angiotensina, antígeno criptocócico, cisticercosis IgG, ADN de
micobacterias, ADN del virus de Epstein-Barr y ADN de borrelia fueron
negativas, al igual que una prueba del Laboratorio de Investigación de
Enfermedades Venéreas. El análisis citológico del LCR mostró una población
mixta de células mononucleares con células plasmáticas raras.
En el octavo día de hospitalización se realizó una
biopsia meníngea. Se inició tratamiento con prednisona a altas dosis, y el
paciente fue dado de alta a su domicilio. Durante las siguientes 2 semanas, el
paciente continuó tomando la prednisona, pero el dolor facial y de cabeza empeoró.
Fue reingresado en el otro hospital para recibir 3 días de tratamiento con
metilprednisolona parenteral en dosis pulsadas.
El examen anatomopatológico de la muestra de biopsia
meníngea reveló una duramadre densamente fibrótica e inflamada crónicamente con
un gran número de macrófagos CD68+ y linfocitos T CD3+ y con colecciones
focales de linfocitos B CD20+. Había un gran número de células plasmáticas
policlonales con más de 100 células plasmáticas IgG4+ por campo de gran
aumento. Había fibrosis estoriforme y la tinción de elastina reveló venulitis
obliterante. Algunos de los focos inflamatorios eran angiocéntricos, pero no
había vasculitis. Había focos raros de necrosis fibrinoide y algunos
neutrófilos, con células gigantes raras pero sin granulomas. Se realizó un
diagnóstico de enfermedad relacionada con IgG4 y se administró rituximab. Dos
días después de la primera dosis de rituximab, se diagnosticó una embolia
pulmonar y se inició tratamiento con apixabán. Dos semanas después, se
repitieron las imágenes para evaluar la respuesta al tratamiento.
La repetición de la resonancia magnética de la cabeza
( Figura 2A y 2B ), realizada después de la administración de contraste intravenoso, reveló un mayor realce
anormal en el seno cavernoso derecho y la fosa craneal media derecha y un
realce anormal nuevo en el seno cavernoso izquierdo y la fosa craneal media
izquierda adyacente. La resonancia magnética de la columna cervical ( Figura 2C
y 2D ) también mostró progresión en la extensión y grosor del engrosamiento y
realce paquimeníngeo ventral y dorsal. También hubo progresión de la estenosis
del canal, que se había vuelto severa, y compresión del cordón a nivel de C7.
No había anormalidad en la señal de la médula.
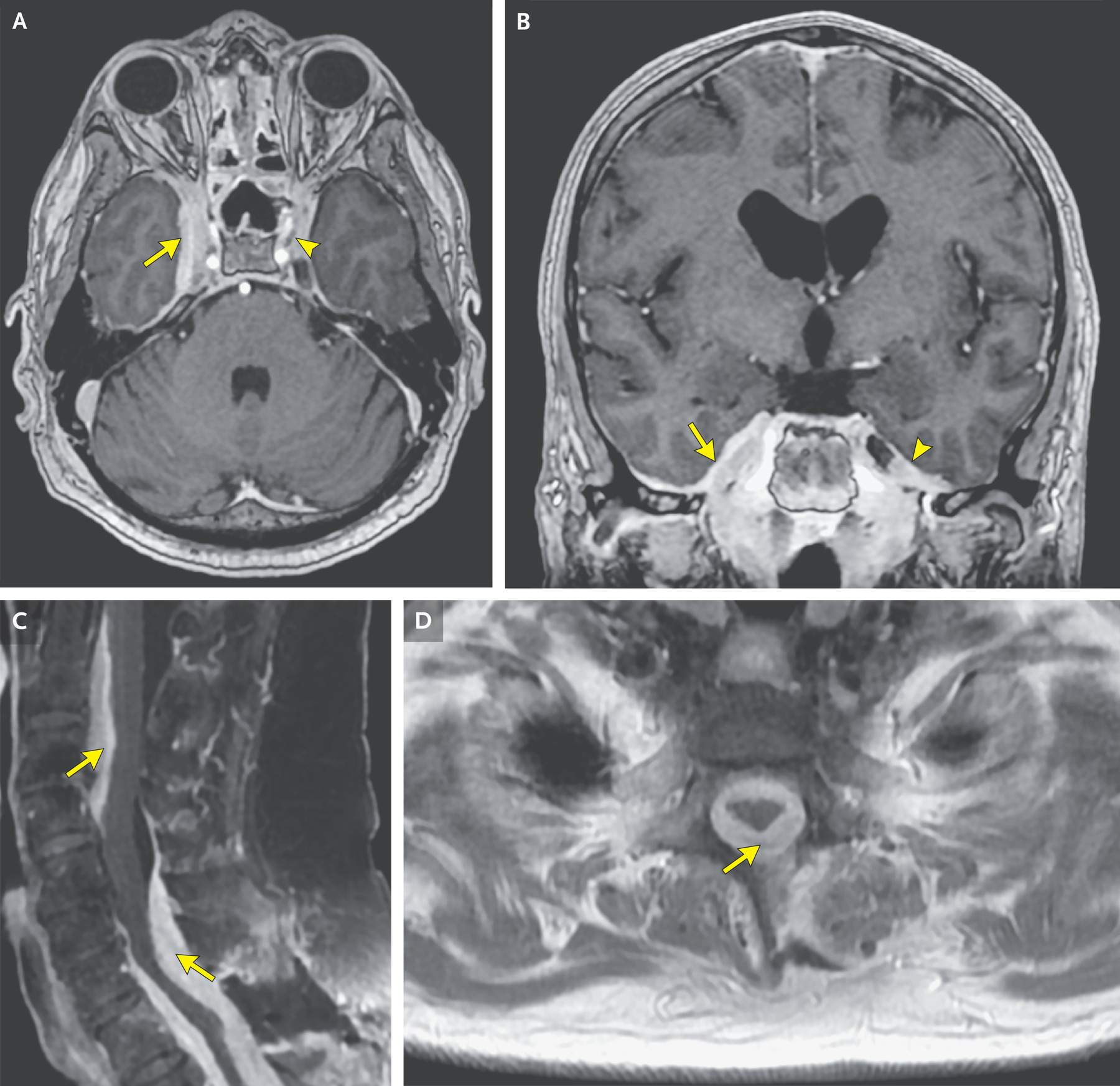
Figura 2. Resonancia magnética de cabeza y columna
obtenida 4 meses antes de la evaluación actual.
Se obtuvo una nueva resonancia magnética de la cabeza
y la columna 2 semanas después de que se iniciara el tratamiento con rituximab.
Las imágenes axiales y coronales potenciadas en T1 con contraste de la cabeza
(Paneles A y B, respectivamente) muestran un aumento anormal del realce en el
lado derecho (flechas) y un realce anormal en el seno cavernoso izquierdo y
fosa craneal media nuevos (puntas de flecha). Imágenes sagitales y axiales
potenciadas en T1 con contraste de la columna cervical con supresión grasa (Paneles C y D,
respectivamente) muestran la progresión del realce paquimeníngeo anormal con empeoramiento
de la estenosis del canal y compresión progresiva de la médula, pero sin
anomalías en la señal de la médula (flechas).
El paciente recibió una segunda infusión de rituximab,
así como tratamiento con dexametasona oral. Tres semanas más tarde, y 7 meses
después del desarrollo de los síntomas iniciales (3 meses antes de la
evaluación actual), el paciente ingresó en el otro hospital debido al
empeoramiento de la cefalea, las náuseas y los vómitos. A la exploración
destacaba hipoacusia neurosensorial de nueva aparición en el oído derecho y
desviación de la lengua hacia la izquierda. Se colocó una derivación
ventriculoperitoneal. Se continuó el tratamiento con dexametasona y el paciente
fue dado de alta a su domicilio. Tres semanas después de la colocación de la
derivación ventriculoperitoneal, el paciente fue ingresado nuevamente en el
otro hospital por infección estreptocócica. Dos meses después del alta y 10
meses después del desarrollo de los síntomas iniciales,
El paciente tenía antecedentes de migrañas,
hipertensión y una mutación heterocigota del factor V de Leiden. Los
medicamentos incluyeron apixabán, baclofeno, dexametasona, furosemida,
gabapentina, lisinopril y metoprolol. Era un empresario jubilado y vivía con su
esposa. Bebía una copa de vino a la semana y había dejado de fumar más de 40
años antes de la evaluación actual. Su abuelo materno había muerto de un
aneurisma aórtico roto.
La temperatura era de 36,5°C, la frecuencia cardíaca
de 88 latidos por minuto, la presión arterial de 118/79 mm Hg y la saturación
de oxígeno del 97% mientras el paciente respiraba aire ambiente. El índice de
masa corporal era de 25,8. El pelo del cuero cabelludo era fino. Tenía facies
cushingoide; acné en la cara, la espalda y el pecho; estrías en los flancos; y
debilidad muscular proximal de brazos y piernas. No había sensibilidad en cuero
cabelludo ni en la región temporal. No tenía lesiones en la lengua ni úlceras
orales, y el examen nasal no mostró costras. Los movimientos extraoculares
estaban intactos. No había artritis. Los exámenes torácico, abdominal y
neurológico fueron normales. El conteo sanguíneo completo y el conteo
diferencial fueron normales, al igual que un análisis de orina. La velocidad de
sedimentación globular fue de 52 mm por hora y el nivel de proteína C reactiva
de 5 mg por litro. La electroforesis de proteínas séricas reveló un nivel de
IgM de 22 mg por decilitro (rango de referencia, 53 a 334) y niveles normales
de IgG e IgA. La prueba de ANCA fue negativa.
Se realizó un diagnóstico y decisiones de manejo.
Diagnóstico diferencial
La enfermedad de este hombre de 59 años comenzó con
fiebre, sudores, rinorrea y dolor maxilar, y presumiblemente fue tratado por
sinusitis. El desarrollo de dolor de cabeza, dolor de mandíbula, sensibilidad
en el cuero cabelludo, polimialgia, diplopía y aumentos marcados en la
velocidad de sedimentación de eritrocitos y el nivel de proteína C reactiva
llevaron a una biopsia de la arteria temporal que condujo a un diagnóstico de
arteritis de células gigantes y tratamiento con prednisona. Después de una
respuesta inicial, los síntomas regresaron cuando la prednisona se redujo
gradualmente a una dosis más baja. A pesar de la adición del tratamiento con
tocilizumab, el dolor de cabeza persistió cuando se redujo gradualmente la
prednisona. Se desarrollaron parálisis de los nervios craneales tercero y
sexto, y se observó que el paciente tenía un título positivo bajo en las
pruebas de ANCA, dirigidas contra la mieloperoxidasa. Se observó evidencia de
paquimeningitis en la resonancia magnética, y una biopsia meníngea reveló
muchas células plasmáticas IgG4+; estos hallazgos provocaron un cambio en el
diagnóstico a enfermedad relacionada con IgG4.
PAQUIMENINGITIS
La paquimeningitis describe una enfermedad que se
caracteriza por el engrosamiento de la duramadre intracraneal, que en la
resonancia magnética puede mostrar realce de gadolinio causado por la
inflamación. La paquimeningitis es una característica central de la
presentación de este paciente; explica el dolor de cabeza, las neuropatías
craneales y la estenosis espinal, y progresó a pesar de la inmunosupresión
sustancial con rituximab y terapia con glucocorticoides. El diagnóstico
diferencial de la paquimeningitis incluye síndromes de vasculitis primaria,
enfermedad inflamatoria crónica no vasculítica, infección y cáncer. La
evaluación de las características extracraneales de la enfermedad de un
paciente y el examen histológico de una muestra de biopsia meníngea son
necesarios para determinar el diagnóstico responsable del síndrome. 1,2
La infección y el cáncer se han descartado en este
paciente sobre la base de estudios microbiológicos e histológicos apropiados, y
no hay características clínicas o histológicas que respalden la histiocitosis.
El diagnóstico de la enfermedad inflamatoria crónica incluye la integración de
las características clínicas y radiológicas con las pruebas serológicas y el
examen anatomopatológico de muestras de biopsia de tejido, así como una
observación cuidadosa a lo largo del tiempo con respecto a la respuesta al
tratamiento. Muchas enfermedades inflamatorias no tienen criterios de
diagnóstico, pero las definiciones de consenso y los sistemas de clasificación
pueden ayudar en el diagnóstico. 3,4 Este paciente había recibido diagnósticos
de dos enfermedades inflamatorias diferentes: arteritis de células gigantes y
enfermedad relacionada con IgG4. Comenzaré considerando cada uno de estos con
más detalle.
ARTERITIS DE CÉLULAS GIGANTES
El dolor de cabeza de este paciente, la sensibilidad
en el cuero cabelludo, el dolor en la mandíbula, la polimialgia y la velocidad
de sedimentación globular y el nivel de proteína C reactiva elevados son
consistentes con el diagnóstico de arteritis de células gigantes. Según los
informes, la biopsia de la arteria temporal reveló evidencia de inflamación
crónica, lo que apoyó aún más el diagnóstico, pero no lo confirmó. Aunque este
paciente cumple con los criterios de clasificación del American College of
Rheumatology de 1990 para la arteritis de células gigantes, no creo que la
arteritis de células gigantes sea el diagnóstico correcto. El curso recurrente
a pesar del tratamiento con tocilizumab y prednisona sería inusual para la
arteritis de células gigantes. Además, los hallazgos de la biopsia de la
arteria temporal incluyeron arteritis necrotizante de un pequeño vaso sanguíneo
adyacente, y la necrosis no es una característica típica de la arteritis de
células gigantes.
ENFERMEDAD RELACIONADA CON IGG4
La enfermedad relacionada con IgG4 tiene una amplia
gama de presentaciones clínicas, y las características histológicas incluyen
fibrosis estoriforme densa con células plasmáticas IgG4+ y venulitis
obliterante. 6 Los niveles circulantes de IgG4 a menudo están elevados. Los
niveles de subclases de IgG fueron normales en este paciente, aunque se
midieron después de meses de tratamiento con terapia con glucocorticoides, lo
que puede haber afectado los resultados. La paquimeningitis es una
característica conocida de la enfermedad relacionada con IgG4. 2 Sin embargo,
la gravedad de la presentación inflamatoria de este paciente, que se
caracterizó por fiebre y sudores nocturnos, sería muy inusual para la
enfermedad relacionada con IgG4, al igual que la mala respuesta del paciente a
los glucocorticoides orales e intravenosos.Los pacientes con enfermedad
relacionada con IgG4 suelen tener una buena respuesta a la prednisona.
Los resultados de las biopsias meníngeas y de la
arteria temporal de este paciente también son útiles cuando se considera el
diagnóstico de enfermedad relacionada con IgG4. Se habría esperado la presencia
de células plasmáticas IgG4+ en la muestra de biopsia de la arteria temporal si
la enfermedad relacionada con IgG4 estuviera afectando la arteria temporal,
pero estas células no se observaron en la muestra de este paciente. 7La biopsia
meníngea reveló fibrosis estoriforme, venulitis obliterante y células
plasmáticas IgG4+, pero estos hallazgos no son diagnósticos de enfermedad
relacionada con IgG4. De hecho, los hallazgos de necrosis fibrinoide en la
biopsia de la arteria temporal y la infiltración de neutrófilos, células
gigantes y necrosis en la biopsia meníngea argumentan fuertemente en contra de
la enfermedad relacionada con IgG4. Cada una de estas características
histológicas es parte de los criterios de exclusión para la enfermedad
relacionada con IgG4 descrita en los criterios de clasificación del American
College of Rheumatology y la European League Against Rheumatism de 2019. 8,9
En cambio, estos hallazgos sugieren la posibilidad de
granulomatosis con poliangeitis (GPA), un tipo de vasculitis asociada a ANCA.
Muchos informes de casos han sugerido una superposición diagnóstica entre la
vasculitis asociada a ANCA y la enfermedad relacionada con IgG4, pero los
estudios observacionales sistemáticos han podido diferenciar entre las
enfermedades. 10 Cuando las características de ambas enfermedades ocurren
simultáneamente, es más probable que el verdadero diagnóstico sea uno u otro,
en lugar de la presencia de ambas enfermedades en el mismo paciente.
GRANULOMATOSIS CON POLIANGEÍTIS
La GPA se ha definido como una inflamación
granulomatosa necrotizante de los vasos sanguíneos de pequeño y mediano tamaño
de las vías respiratorias que, a menudo, también afecta a los riñones. 11 Las
características inflamatorias observadas en este paciente son consistentes con
este diagnóstico, y tanto el dolor facial como la sensibilidad maxilar también
son comunes con la GPA. La ausencia de hallazgos en los pulmones y los riñones
haría de este un caso inusual de GPA, pero tales casos ocurren. Las
presentaciones que se limitan a la cabeza y el cuello, incluida la paquimeningitis
hipertrófica, están bien descritas en pacientes con GPA.
El GPA está estrechamente relacionado con uno de los
dos autoanticuerpos dirigidos contra los antígenos citoplásmicos de los
neutrófilos: la proteinasa 3 o la mieloperoxidasa. Este paciente tuvo una
prueba positiva de anticuerpos contra la mieloperoxidasa, aunque a un nivel
relativamente bajo. Los ensayos de ANCA siguen estando poco estandarizados y su
rendimiento diagnóstico depende tanto de la probabilidad de enfermedad previa a
la prueba como de la experiencia del laboratorio. 12 Además, el título en este
paciente puede haberse reducido en las 6 semanas anteriores de tratamiento con
prednisona.
La inflamación necrosante de la pequeña arteria que se
observó en la biopsia de la arteria temporal y la presencia de células gigantes
con áreas de necrosis en la biopsia meníngea son compatibles con GPA. Además, a
menudo se detectan infiltraciones de células plasmáticas IgG4+ en esta
enfermedad. 13 GPA inicialmente responde a los glucocorticoides, pero la
remisión requiere tratamiento con el fármaco citotóxico ciclofosfamida o el
anticuerpo que agota las células B, rituximab. El rituximab tarda en hacer
efecto y aún no habría sido efectivo dentro de las 2 semanas posteriores al
tratamiento, cuando los hallazgos radiológicos de este paciente habían
progresado y se había producido un deterioro clínico.
Diagnóstico presuntivo
Granulomatosis con poliangeítis.
Discusión patológica
Se revisaron las muestras de las biopsias meníngeas y
de la arteria temporal que se realizaron en el otro hospital. La muestra de
biopsia de la arteria temporal ( Figura 3 ) mostró vasculitis necrosante que
afectaba a una rama de la arteria. La arteria temporal principal no estaba
involucrada. Había tanto necrosis fibrinoide como trombosis luminal de la rama
arterial. También se observó afectación focal de pequeñas venas, lo que indica
una vasculitis de pequeños vasos. La tinción inmunohistoquímica mostró que el
infiltrado inflamatorio estaba compuesto principalmente por linfocitos T CD3+,
células plasmáticas CD138+ y macrófagos CD68+.

Figura 3. Muestra de biopsia de arteria temporal.
La tinción con hematoxilina y eosina (Panel A) muestra
inflamación (flechas) con necrosis fibrinoide (punta de flecha) y trombosis luminal
(asterisco) que afecta a una rama de la arteria; la arteria temporal principal
a la izquierda no está afectada. A mayor aumento (Panel B), se puede ver la
afectación focal de una vena pequeña, lo que indica una vasculitis de vasos
pequeños (flecha). Cortes seriados a mayor aumento (Paneles C a F) muestran
vasculitis necrotizante de una arteria pequeña en la tinción de hematoxilina y
eosina (Panel C, punta de flecha), con las células inflamatorias compuestas de
linfocitos T, resaltado por una tinción inmunohistoquímica para CD3 (Panel D,
en marrón); células plasmáticas, destacada por un inmunotinción histoquímica
para CD138 (Panel E, en marrón); y macrófagos, resaltados por una tinción
inmunohistoquímica para CD68 (Panel F, en marrón).
Aunque la biopsia de la arteria temporal mostró
vasculitis, y la arteritis de células gigantes es, con mucho, la forma más
común de vasculitis que afecta este sitio, hasta en el 6% de las biopsias de la
arteria temporal que confirman la presencia de vasculitis, la vasculitis se
debe a una forma distinta de la arteritis de células gigantes. 14-16Debe
evitarse el uso del término “arteritis temporal” para describir las biopsias de
la arteria temporal con evidencia de vasculitis porque esta construcción
inadvertidamente implica arteritis de células gigantes. Varias características
histológicas de la muestra de biopsia de la arteria temporal de este paciente
eran inusuales para la arteritis de células gigantes: el hallazgo de arteritis
confinada principalmente a una rama de la arteria, la presencia de necrosis
fibrinoide y trombosis luminal, y el grado de infiltración de células
plasmáticas. Además, la afectación venosa implica una vasculitis de vasos
pequeños (p. ej., una vasculitis relacionada con ANCA o una vasculitis mediada
por inmunocomplejos). 17 Aunque la enfermedad relacionada con IgG4 puede estar
asociada con vasculitis tanto en vasos pequeños como grandes, la vasculitis
necrosante de vasos pequeños no es una característica de la enfermedad
relacionada con IgG4.7,18 Tomados en conjunto, los hallazgos de la biopsia de
la arteria temporal no fueron consistentes con arteritis de células gigantes ni
con enfermedad relacionada con IgG4.
La muestra de biopsia meníngea mostró un infiltrado
inflamatorio difuso similar al observado en la muestra de biopsia de arteria
temporal; el infiltrado estaba compuesto principalmente por linfocitos T CD3+,
células plasmáticas CD138+ y macrófagos CD68+ ( Figura 4). Relativamente pocos
linfocitos B CD20+ estaban presentes, y las células plasmáticas mostraron una
expresión politípica para las cadenas ligeras de inmunoglobina kappa y lambda
(no mostrado). Había células gigantes multinucleadas ocasionales, que indicaban
inflamación granulomatosa, así como áreas con neutrófilos y necrosis (a menudo
denominadas microabscesos). No se identificaron la fibrosis estoriforme y la
venulitis obliterante, que se informaron en el otro hospital y se pueden ver en
algunos casos de enfermedad relacionada con IgG4. La tinción para
microorganismos fue negativa. Había más de 100 células plasmáticas IgG4+ por
campo de alta potencia; El 49% de las células plasmáticas IgG+ mostraron
expresión de IgG4. La naturaleza difusa del infiltrado meníngeo no era
compatible con una vasculitis focalizada como la arteritis de células gigantes.
El grado de infiltración de células plasmáticas IgG4+ cumpliría los criterios
de enfermedad relacionada con IgG4 en el contexto apropiado; sin embargo, las
células gigantes y los microabscesos neutrofílicos no son características de la
enfermedad relacionada con IgG4. 18 La inflamación difusa con células gigantes
y microabscesos neutrofílicos son características de la GPA. 19 Las muestras de
biopsia de pacientes con GPA también pueden mostrar un alto grado de
infiltración tisular por células plasmáticas IgG4+. 20 En conjunto, este caso
de vasculitis que afecta tanto a la arteria temporal como a las meninges es más
consistente con el diagnóstico de GPA.

Figura 4. Muestra de biopsia meníngea.
La tinción con hematoxilina y eosina (Panel A) muestra
inflamación (flechas). Las células inflamatorias están compuestas de linfocitos
T, resaltado por inmunohistoquímica para CD3 (Panel B, en marrón); las células
plasmáticas resaltadas por una tinción inmunohistoquímica para CD138 (Panel C,
en marrón); y macrófagos, destacados por una tinción inmunohistoquímica para
CD68 (Panel D, en marrón). A mayor aumento en inmunohistoquimica, las células
plasmáticas muestran una fuerte expresión de IgG (Panel E, en marrón), con 49%
del plasma IgG+ células que muestran expresión de IgG4 (Panel F, en marrón). A
mayor aumento en la tinción de hematoxilina y eosina, células gigantes raras (Panel
G, flecha) y se ven microabscesos neutrofílicos (Panel H, flecha).
DIAGNÓSTICO PATOLÓGICO
ARTERITIS QUE AFECTA A LA ARTERIA TEMPORAL Y
PAQUIMENINGITIS ASOCIADA A GRANULOMATOSIS CON POLIANGITIS.
Discusión de manejo
El tratamiento de la GPA generalmente requiere al
menos dos terapias farmacológicas, no solo glucocorticoides, para inducir la
remisión. 11 Este paciente había recibido una segunda dosis de rituximab solo 3
meses antes de ser atendido en la consulta de reumatología de este hospital.
Durante el tiempo intermedio, se le había colocado una derivación
ventriculoperitoneal que se complicó con una infección estreptocócica. Aunque
su estado parecía estable en la clínica de reumatología el día que nos
conocimos, la velocidad de su deterioro neurológico anterior sugirió que el
control de la enfermedad era tenue y que se justificaba la terapia de
consolidación. El examen físico destacó los efectos de muchos meses de terapia
con glucocorticoides, 21incluyendo facies cushingoide, cabello fino, múltiples
signos cutáneos de efectos tóxicos y miopatía inducida por glucocorticoides.
Éramos reacios a mantener o intensificar su nivel actual de tratamiento con
glucocorticoides o a agregar otro medicamento como la ciclofosfamida en vista
de la infección reciente del paciente.
Optamos por aumentar el agotamiento de las células B
en este paciente como un enfoque para consolidar el control de la enfermedad.
Las células B circulantes representan solo del 3 al 5% de la reserva total de
linfocitos B de una persona. Aunque las células B circulantes ya no se detectan
incluso después de una sola dosis de 1000 mg de rituximab en la mayoría de los
casos, estos pacientes no están realmente "agotados de células B". La
capacidad para medir la profundidad del agotamiento de las células B con el uso
de citometría de flujo convencional es deficiente, y los ensayos que están
disponibles actualmente para fines clínicos de rutina no brindan una indicación
precisa de la medida en que se ha visto afectada la reserva de linfocitos B.
Las decisiones sobre cuándo volver a administrar agentes que agotan las células
B son en gran medida empíricas. Para este paciente con enfermedad
particularmente refractaria a la inducción de remisión, administramos una
tercera dosis de rituximab, seguida de una cuarta dosis 4 meses después. Con
este régimen se logró un excelente control de la enfermedad. Los
glucocorticoides se redujeron después de varios meses y permanece en remisión
completa bajo estrecha vigilancia. Sin embargo, más de 1 año después de que se
logró la remisión y se suspendieron por completo los glucocorticoides, el
paciente comenzó a tener necrosis avascular de ambas caderas y el hombro
izquierdo, complicaciones a largo plazo de la terapia intensiva con
glucocorticoides utilizada al principio de su curso clínico.
DIAGNOSTICO FINAL
GRANULOMATOSIS CON POLIANGEÍTIS.
Traducción de:
Case 28-2022: A 59-Year-Old Man with Headache and
Progressive Neurologic Dysfunction
David Jayne, M.D., John H. Stone, M.D., M.P.H., Otto
Rapalino, M.D., and James R. Stone, M.D., Ph.D.
https://www.nejm.org/doi/full/10.1056/NEJMcpc2100271
Referencias
1. Mekinian A, Maisonobe L, Boukari L,
et al. Characteristics, outcome and treat[1]ments
with cranial pachymeningitis: a mul[1]ticenter
French retrospective study of 60
patients. Medicine (Baltimore) 2018;97(30):
e11413.
2. Wallace ZS, Carruthers MN, Khosro[1]shahi
A, et al. IgG4-related disease and
hypertrophic pachymeningitis. Medicine
(Baltimore) 2013;92:206-16.
3. Jennette JC, Falk RJ, Bacon PA, et al.
2012 Revised International Chapel Hill
Consensus Conference nomenclature of
vasculitides. Arthritis Rheum 2013;65:1-11.
4. Aringer M, Costenbader K, Daikh D,
et al. 2019 European League Against
Rheumatism/American College of Rheu[1]matology
classification criteria for sys[1]temic
lupus erythematosus. Ann Rheum
Dis 2019;78:1151-9.
5. Hunder GG, Bloch DA, Michel BA, et al.
The American College of Rheumatology
1990 criteria for the classification of giant
cell arteritis. Arthritis Rheum 1990;33:
1122-8.
6. Perugino CA, Stone JH. IgG4-related
disease: an update on pathophysiology
and implications for clinical care. Nat Rev
Rheumatol 2020;16:702-14.
7. Monach PA, Stone JH, Sharma A,
Nazarian RM. Case 6-2017: a 57-year-old
woman with fatigue, sweats, weight loss,
headache, and skin lesions. N Engl J Med
2017;376:775-86.
8. Wallace ZS, Zhang Y, Perugino CA,
et al. Clinical phenotypes of IgG4-related
disease: an analysis of two international
cross-sectional cohorts. Ann Rheum Dis
2019;78:406-12.
9. Wallace ZS, Naden RP, Chari S, et al.
The 2019 American College of Rheuma[1]tology/European
League Against Rheuma[1]tism
classification criteria for IgG4-related
disease. Ann Rheum Dis 2020;79:77-87.
10. Erden A, Bolek EC, Yardimci KG, Kilic
L, Bilgen SA, Karadag O. Do ANCA-asso[1]ciated
vasculitides and IgG4-related dis[1]ease
really overlap or not? Int J Rheum Dis
2019;22:1926-32.
11. Hoffman GS, Kerr GS, Leavitt RY, et al.
Wegener granulomatosis: an analysis of
158 patients. Ann Intern Med 1992;116:
488-98.
12. Bossuyt X, Cohen Tervaert J-W,
Arimura Y, et al. Position paper: revised
2017 international consensus on testing
of ANCAs in granulomatosis with poly[1]angiitis
and microscopic polyangiitis. Nat
Rev Rheumatol 2017;13:683-92.
13. Danlos F-X, Rossi GM, Blockmans D,
et al. Antineutrophil cytoplasmic antibody[1]associated
vasculitides and IgG4-related
disease: a new overlap syndrome. Auto[1]immun
Rev 2017;16:1036-43.
14. Généreau T, Lortholary O, Pottier MA,
et al. Temporal artery biopsy: a diagnostic
15. Hamidou MA, Moreau A, Toquet C, El
Kouri D, de Faucal P, Grolleau J-Y. Tempo[1]ral
arteritis associated with systemic nec[1]rotizing
vasculitis. J Rheumatol 2003;30:
2165-9.
16. Cavazza A, Muratore F, Boiardi L, et al.
Inflamed temporal artery: histologic find[1]ings
in 354 biopsies, with clinical correla[1]tions.
Am J Surg Pathol 2014;38:1360-70.
17. Jennette JC, Stone JR. Chapter 11 —
diseases of medium-sized and small ves[1]sels.
In: Willis MS, Homeister JW, Stone
JR, eds. Cellular and molecular pathobiol[1]ogy
of cardiovascular disease. San Diego,
CA: Academic Press, 2014;197-219 (http://
www.sciencedirect.com/science/article/pii/
B9780124052062000119).
18. Deshpande V, Zen Y, Chan JK, et al.
Consensus statement on the pathology of
IgG4-related disease. Mod Pathol 2012;
25:1181-92.
19. Merkel PA, McCarty D, Sharma A,
Stone JR. Case 31-2008: a 39-year-old man
with chest pain, arthralgias, and a medi[1]astinal
mass. N Engl J Med 2008;359:
1603-14.
20. Chang SY, Keogh KA, Lewis JE, et al.
IgG4-positive plasma cells in granulomato[1]sis
with polyangiitis (Wegener’s): a clini[1]copathologic
and immunohistochemical
study on 43 granulomatosis with polyan[1]giitis
and 20 control cases. Hum Pathol
2013;44:2432-7.
21. McDowell PJ, Stone JH, Zhang Y, et al.
Quantification of glucocorticoid-asso[1]ciated
morbidity in severe asthma using
the glucocorticoid toxicity index. J Al[1]lergy
Clin Immunol Pract 2021;9(1):365-
372.e5.
No hay comentarios:
Publicar un comentario