Un hombre de 33 años de edad fue evaluado en la
clínica de gastroenterología de este hospital debido a una diarrea crónica
recurrente y un diagnóstico de enteropatía autoinmune.
La diarrea se había desarrollado inicialmente en el
paciente durante la infancia. Cuando el paciente tenía 2 meses de edad, tenía
un crecimiento deficiente y deposiciones frecuentes con heces blandas. Cuando
comenzó a comer alimentos sólidos, la diarrea continuó y se inició una dieta de
proteínas hidrolizadas. Sus padres recibieron instrucciones de eliminar los
lácteos, la soya y el gluten de su dieta, pero la diarrea no disminuyó. Las
pruebas cutáneas de alergia con antígenos de leche de vaca y soja fueron
negativas.
Cuando el paciente tenía 10 meses de edad, persistía
el retraso del crecimiento y la diarrea, y se desarrolló edema generalizado y
síndrome nefrótico. Se realizó una esofagogastroduodenoscopia (EGD) y, según
los informes, el examen histopatológico de una muestra de biopsia del intestino
delgado reveló atrofia de las vellosidades, hiperplasia de las criptas e
inflamación difusa. Según los informes, la microscopía electrónica de
transmisión mostró cambios degenerativos leves del epitelio superficial y
atrofia microvellosa focal leve; no había depósitos inmunes y la membrana basal
era normal.
También se realizó una biopsia renal y el examen
histopatológico de una muestra de biopsia renal reveló glomérulos con
engrosamiento difuso de la membrana basal; no hubo proliferación de células o
matriz mesangial. La microscopía electrónica de transmisión mostró borramiento
total de los procesos de los pies de las células epiteliales y una membrana
basal glomerular irregularmente engrosada con numerosos depósitos
subepiteliales e intramembranosos densos en electrones. El paciente recibió
diagnóstico de síndrome nefrótico por glomerulonefritis membranosa y se inició
tratamiento con prednisona.
La diarrea disminuyó, la proteinuria y el edema
generalizado se resolvieron y se reanudó el crecimiento. Sin embargo, durante
los siguientes 2 años, se produjeron recaídas intermitentes de diarrea y
síndrome nefrótico cuando se disminuyó la dosis de prednisona. Cuando el
paciente tenía 3 años de edad, se inició tratamiento con clorambucilo por
glomerulonefritis membranosa recurrente, y no hubo recaídas durante 1 año.
Cuando el paciente tenía 4 años de edad, presentó
recurrencia de diarrea asociada con fiebre y anemia hemolítica. El cultivo de
heces, la prueba de heces para la toxina de Clostridioides difficile y el
examen de las heces para huevos y parásitos fueron negativos. Se realizó EGD y,
según los informes, el examen histopatológico de una muestra de biopsia de
intestino delgado reveló atrofia de las vellosidades, hiperplasia de las
criptas y enteritis activa. El nivel de IgE en sangre fue de 501 UI por
mililitro (rango de referencia, 0 a 100). El nivel en sangre de anticuerpos
contra el endomisio no se elevó, pero se detectaron anticuerpos anticélulas
epiteliales. El paciente recibió el diagnóstico de enteropatía autoinmune y se
inició tratamiento con glucocorticoides intravenosos y ciclosporina.
Después de varios meses, se suspendió el tratamiento
con glucocorticoides y ciclosporina y se inició tacrolimus. El tacrolimus se
suspendió después de varios años, cuando los síntomas del paciente se
resolvieron. De los 5 a los 13 años no presentó enteropatía, anemia ni
nefropatía.
A partir de los 13 años de edad, el paciente fue
hospitalizado por recurrencias de diarrea y nefropatía cada 2 a 4 años durante
los 20 años anteriores a esta evaluación. Seis años antes de esta evaluación,
se realizó EGD y colonoscopia, y la mucosa duodenal y colónica aparecía
difusamente edematosa. Se obtuvieron muestras de biopsia.
El examen de una muestra de biopsia duodenal ( Figura
1 ) reveló un marcado embotamiento de las vellosidades, un aumento de células
inflamatorias linfoplasmocíticas en la lámina propia (conocida como expansión
linfoplasmocítica de la lámina propia) y un aumento irregular de linfocitos
intraepiteliales. El examen de una muestra de biopsia colónica reveló una
colitis activa focal leve que no afectaba al recto.

Figura 1. Muestras de biopsia de intestino delgado.
En la tinción con hematoxilina y eosina, una muestra
de mucosa duodenal obtenida 6 años antes de esta presentación (Panel A) muestra
un marcado embotamiento de las vellosidades (flechas) y expansión linfoplasmocítica
de la lámina propia. Una muestra de la mucosa duodenal obtenida en la
presentación (Paneles B y C) muestra un marcado embotamiento de las vellosidades
(Panel B, flechas), expansión linfoplasmocítica de la lámina propia y un
aumento de linfocitos intraepiteliales (Paneles B y C, asteriscos). Una muestra
de mucosa ileal obtenida en la presentación (Panel D) muestra hallazgos similares
a los observados en el duodeno, con embotamiento de las vellosidades leves
(flechas), expansión linfoplasmocítica de la lámina propia y un aumento de
linfocitos intraepiteliales (asteriscos)
Se inició tratamiento con metilprednisolona
intravenosa y disminuyó la diarrea. Durante los siguientes 2 años, las recaídas
de diarrea y síndrome nefrótico se trataron con metilprednisolona intravenosa o
prednisona oral en combinación con ciclosporina, tacrolimus y micofenolato
mofetilo.
Cuatro años antes de esta evaluación, no se detectaron
anticuerpos contra el receptor de fosfolipasa A 2 de tipo M. Se realizó una
biopsia renal y el examen histopatológico de una muestra de biopsia renal
nuevamente reveló evidencia de glomerulonefritis membranosa. Se cambió el
tratamiento a prednisona, budesonida y abatacept, y los síntomas remitieron.
Después de 2 años, el paciente dejó de tomar abatacept. Después de otros 2
años, la diarrea volvió a aparecer y el paciente fue derivado a la clínica de
gastroenterología de este hospital.
En la consulta de gastroenterología se obtuvo la
anamnesis adicional. Había antecedentes de hipertensión, cálculos biliares y
rosácea. El eczema se había desarrollado cuando el paciente tenía 10 meses de
edad y había ocurrido de forma intermitente hasta la edad adulta; había
disminuido con la administración de agentes inmunosupresores. Un episodio de
shock anafiláctico había ocurrido cuando el paciente tenía 2 años de edad
después de haber estado expuesto a huevo crudo. En ese momento, se realizaron
pruebas de alergia y se indicó a los padres del paciente que eliminaran los
huevos, los frutos secos, los cacahuetes, el pescado y los mariscos de su
dieta, además de eliminar los lácteos, la soja y el gluten.
Los medicamentos actuales incluían prednisona, budesonida
y losartán. No se conocían alergias a medicamentos. El paciente vivía en un
estado del Atlántico Medio y trabajaba como vendedor. No fumaba tabaco y rara
vez bebía cerveza. Su historial familiar incluía cáncer de ovario en su madre y
cáncer de piel en su padre; su hermano estaba sano.
En el examen, la temperatura era de 37,0°C, la presión
arterial de 131/94 mm Hg, la frecuencia cardíaca de 106 latidos por minuto y la
saturación de oxígeno del 97% mientras el paciente respiraba aire ambiente. El
índice de masa corporal fue de 24,1. El paciente presentaba baja estatura y
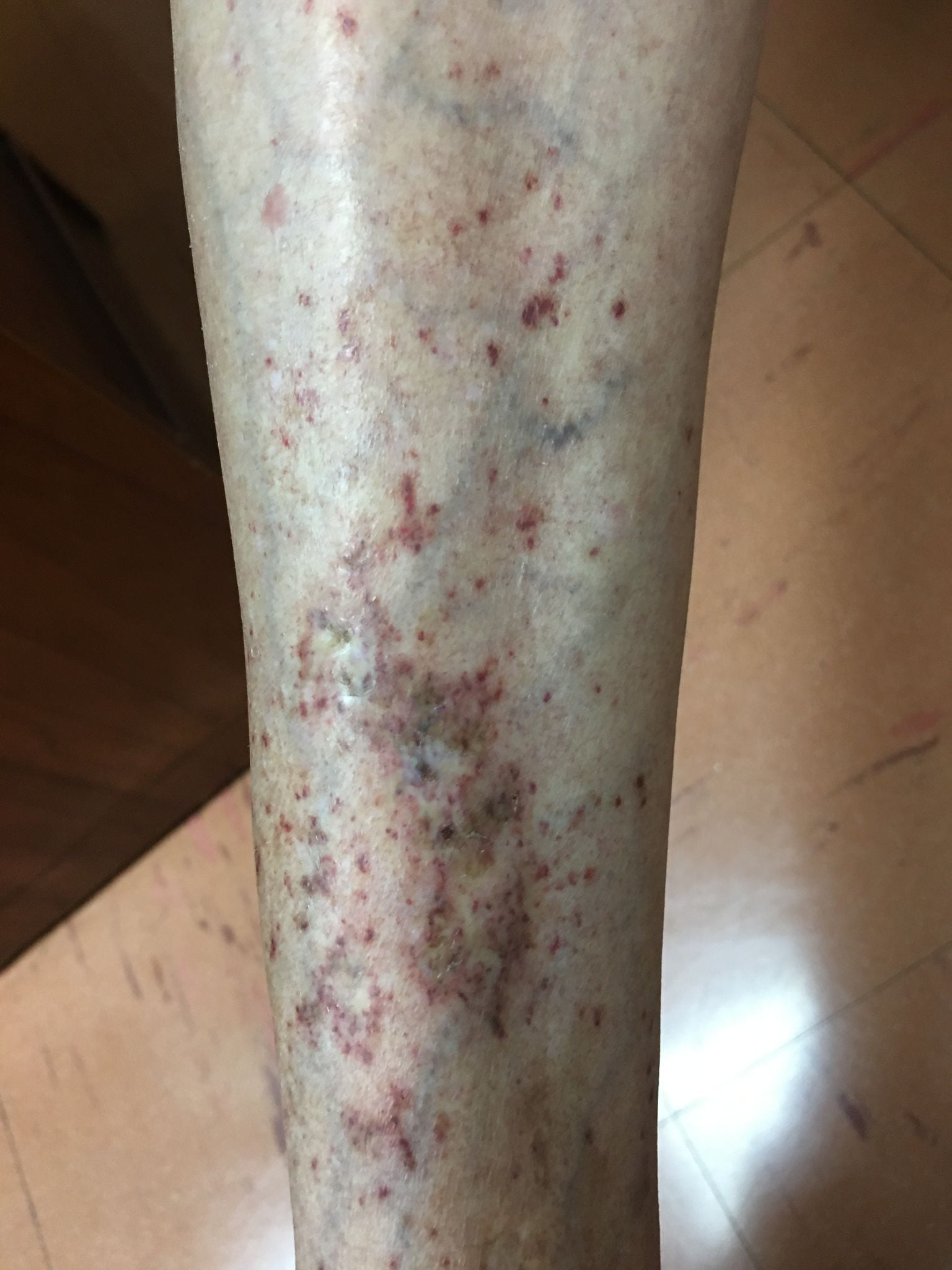
eritema difuso en cara y cuello. Había sensibilidad leve en la parte inferior
izquierda del abdomen. Los resultados de una prueba de aliento con lactulosa
fueron normales, al igual que el nivel de calprotectina fecal. La prueba de
anticuerpos contra la transglutaminasa tisular fue negativa. En la Tabla 1 se
muestran otros resultados de pruebas de laboratorio . Se realizó EGD y
colonoscopia, y los hallazgos fueron compatibles con duodenitis, gastritis y
atrofia de colon. Se obtuvieron muestras de biopsia.


Tabla 1. Datos de laboratorio.
El examen histopatológico de la biopsia ileal y
duodenal ( Figura 1 ) reveló embotamiento de las vellosidades, expansión
linfoplasmocitaria de la lámina propia y linfocitosis intraepitelial.
Se realizó una prueba de diagnóstico.
Diagnóstico diferencial
Este hombre de 33 años tenía un diagnóstico de larga
data de enteropatía autoinmune, que se había complicado con anemia hemolítica,
nefropatía membranosa con proteinuria y eczema. Los síntomas se controlaron
intermitentemente con la administración de glucocorticoides y otros agentes
inmunosupresores, pero nunca se logró una remisión completa y duradera. ¿La
presentación actual del paciente con diarrea recurrente podría deberse a la
persistencia o exacerbación de su afección primaria, la enteropatía autoinmune?
¿O podría ser causado por otra condición que adquirió debido a la
inmunosupresión crónica? Alternativamente, ¿podría su diagnóstico de
enteropatía autoinmune, que había llevado durante la mayor parte de su vida,
ser incorrecto? El primer paso en el cuidado de este paciente es reconsiderar
el diagnóstico diferencial de diarrea crónica y malabsorción que pueden ocurrir
en asociación con atrofia de las vellosidades y enteropatía.
ENTEROPATÍA AUTOINMUNE
La enteropatía autoinmune es una condición rara que es
más común entre bebés y niños que entre adultos. Puede ocurrir como un proceso
primario o deberse a otro proceso inmunomediado. Los síntomas incluyen diarrea
crónica, malabsorción y pérdida de peso. Las manifestaciones extraintestinales
pueden incluir anemia hemolítica, nefritis y dermatitis, todas las cuales
habían ocurrido en este paciente. También se han informado tiroiditis y
hepatitis como manifestaciones extraintestinales de enteropatía autoinmune.
Los criterios diagnósticos propuestos para la
enteropatía autoinmune incluyen la detección de atrofia de las vellosidades en
la biopsia del intestino delgado, junto con una disminución de las células
caliciformes en el tejido; la presencia de anticuerpos antienterocitos; y
diarrea crónica con malabsorción. 1 En este paciente se detectaron anticuerpos
anticélulas epiteliales cuando tenía 4 años. En ese momento, la prueba de anticuerpos
antienterocitos aún no estaba disponible y el paciente cumplía con los
criterios para el diagnóstico de enteropatía autoinmune. Cuando evaluamos
inicialmente a este paciente, la prueba sérica de anticuerpos antienterocitos
fue positiva con un título de 1:10. Sin embargo, se consideraron posibles
causas distintas a la enteropatía autoinmune, dada la naturaleza refractaria de
la diarrea y los otros síntomas clínicos sustanciales.
ENFERMEDAD CELÍACA
Los pacientes con enfermedad celíaca también tienen diarrea,
malabsorción y pérdida de peso. Las manifestaciones extraintestinales pueden
incluir nefropatía por IgA y dermatitis herpetiforme, ninguna de las cuales
ocurrió en este paciente. Además, la enfermedad celíaca no suele asociarse con
anemia hemolítica, 2 que había sido una característica destacada del curso de
la enfermedad de este paciente.
En pacientes con enfermedad celíaca, la evaluación
serológica confirma la presencia de transglutaminasa tisular IgA; si el
paciente tiene una deficiencia selectiva de IgA, se puede medir la
transglutaminasa tisular IgG. En este paciente, las pruebas serológicas para
transglutaminasa tisular IgA fueron negativas en ausencia de deficiencia de
IgA. Además, la enfermedad celíaca se trata con la eliminación del gluten de la
dieta y los síntomas de este paciente no disminuyeron con la eliminación del
gluten en la dieta. El esprúe colágeno es un subtipo raro de enfermedad celíaca
que a menudo es refractario al tratamiento convencional, pero se caracteriza
por la presencia de una banda subepitelial de colágeno en el examen
histopatológico de una muestra de biopsia del intestino delgado, hallazgo que
no se observó en este paciente. .
ENFERMEDAD INFLAMATORIA INTESTINAL
La enfermedad inflamatoria intestinal puede causar diarrea
y malabsorción, pero rara vez se desarrolla en la infancia. Sin embargo, se ha
reportado una entidad descrita como enfermedad inflamatoria intestinal de
inicio muy temprano; se ha asociado con ciertas variantes genéticas. 3 Aunque
la enfermedad inflamatoria intestinal no suele asociarse con manifestaciones
renales, se han informado casos de nefritis intersticial granulomatosa y
nefropatía IgA en algunos pacientes con enfermedad inflamatoria intestinal. 4,5
Este paciente no tenía evidencia de enfermedad inflamatoria intestinal en la
endoscopia y su nivel de calprotectina fecal era normal.
CRECIMIENTO EXCESIVO DE BACTERIAS EN EL INTESTINO
DELGADO
El crecimiento excesivo de bacterias en el intestino
delgado es una afección caracterizada por distensión abdominal, diarrea y
calambres. Es causada por una colonización excesiva del intestino delgado con
microbios colónicos y se trata con agentes antibióticos orales. Este paciente
no tenía condiciones subyacentes confirmadas que se sabe que confieren una predisposición
al crecimiento excesivo de bacterias en el intestino delgado, como enfermedad
inflamatoria intestinal, divertículos en el intestino delgado, resecciones
intestinales, trastornos de la motilidad, diabetes, esclerodermia o deficiencia
de IgA. Además, sus manifestaciones extraintestinales no se explicarían por un
sobrecrecimiento bacteriano del intestino delgado. Una prueba de aliento con
lactulosa fue negativa, lo que argumenta en contra de este diagnóstico, pero
debido a que las pruebas de aliento con carbohidratos tienen una sensibilidad y
especificidad variables, este hallazgo no descarta el sobrecrecimiento
bacteriano en el intestino delgado.
INTOLERANCIA A LA LECHE DE VACA
La intolerancia a la leche de vaca es común entre los
bebés y los niños pequeños. Provoca diarrea y puede asociarse con eccema. La
intolerancia a la leche de vaca generalmente se resuelve con la edad, mientras
que los síntomas de este paciente persistieron hasta la edad adulta. Además, la
prueba de punción cutánea con antígeno de leche de vaca fue negativa y los
síntomas persistieron a pesar de la eliminación de la leche de vaca de la
dieta.
GASTROENTERITIS EOSINOFÍLICA
La gastroenteritis eosinofílica es más frecuente entre
los niños menores de 5 años y a menudo se asocia con una afección atópica
coexistente. Debido a que suele haber un componente alérgico subyacente, puede
haber eosinofilia y un nivel elevado de IgE en sangre. El nivel de IgE en
sangre a veces estaba elevado en este paciente, y la ausencia de eosinofilia podría
explicarse por el tratamiento en curso con glucocorticoides. Sin embargo, no
hubo evidencia de compromiso tisular con infiltración eosinofílica en la mucosa
o capas musculares en múltiples biopsias del intestino delgado.
ENTEROPATÍA ASOCIADA CON BLOQUEADORES DE LOS
RECEPTORES DE ANGIOTENSINA
Se ha notificado enteropatía tipo sprue en pacientes
que toman el bloqueador del receptor de la angiotensión II olmesartán. Ha
habido muy pocos informes de casos de esta afección con el uso de losartán, 6
que era el bloqueador de los receptores de angiotensión II que estaba tomando
este paciente. Los síntomas de este paciente eran anteriores a su uso de
losartán, por lo que es poco probable que este medicamento sea la causa de sus
síntomas; sin embargo, sería apropiado un ensayo breve que implique la
interrupción de este medicamento.
ERRORES INNATOS DE LA INMUNIDAD
Los errores congénitos de la inmunidad, también
conocidos como inmunodeficiencias primarias, son trastornos que se derivan de
defectos en la tolerancia o la regulación inmunitaria y pueden simular una
enfermedad inflamatoria intestinal. Muchas de estas condiciones pueden ser
fatales en los primeros años de vida. La inmunodeficiencia primaria más común,
la inmunodeficiencia común variable, se puede descartar en este paciente, dada
la ausencia de deficiencias de inmunoglobulinas. Dos inmunodeficiencias
primarias adicionales merecen consideración en este paciente:
poliendocrinopatía autoinmune, candidiasis y distrofia ectodérmica (APECED) y
desregulación inmune, poliendocrinopatía, enteropatía, síndrome ligado al
cromosoma X (IPEX).
APECED
APECED, también conocido como síndrome poliendocrino
autoinmune tipo 1 (APS-1), resulta de un defecto en el gen regulador autoinmune
( AIRE ) que conduce a una pérdida de la tolerancia inmunitaria central. 7 Los
pacientes con APECED presentan candidiasis mucocutánea, distrofia ectodérmica y
múltiples enfermedades endocrinas. El hipoparatiroidismo y la insuficiencia
suprarrenal son las enfermedades endocrinas más comunes; también puede ocurrir
diabetes mellitus o hipotiroidismo (o ambos). APECED a menudo se asocia con
enteropatía autoinmune subyacente y las manifestaciones gastrointestinales
pueden variar ampliamente. Sin embargo, este paciente no tenía ninguna de las
enfermedades endocrinas típicas asociadas con APECED ni candidiasis
mucocutánea.
Síndrome IPEX
El síndrome IPEX es un trastorno de desregulación
inmunitaria que normalmente se desarrolla en la infancia. Se debe a un defecto
en el gen que codifica la proteína de caja de cabeza de horquilla 3 ( FOXP3 )
que conduce a células T reguladoras disfuncionales. 8 Los pacientes con
síndrome IPEX presentan una tríada clásica de manifestaciones: endocrinopatías
autoinmunes (típicamente diabetes mellitus tipo 1 neonatal y tiroiditis),
enteropatía autoinmune (asociada con retraso del crecimiento y diarrea crónica
grave) y dermatitis eccematosa.La anemia hemolítica y la nefritis, que se
observaron en este paciente, se han descrito en pacientes con síndrome IPEX. La
biopsia intestinal típicamente revela características compatibles con
enteropatía autoinmune: atrofia de las vellosidades, linfocitos
intraepiteliales y células plasmáticas. La terapia inmunosupresora es el pilar
del tratamiento. Sin embargo, la inmunosupresión generalmente controla los
síntomas solo de manera transitoria, sin remisión a largo plazo, lo cual es
consistente con el curso de la enfermedad de este paciente.
Dada la historia de este paciente, la presentación
clínica y la constelación de síntomas, sospeché que el síndrome IPEX era el
diagnóstico más probable. Para establecer este diagnóstico, remití al paciente
a la unidad de alergia e inmunología clínica para una evaluación adicional y
consideración para pruebas genéticas.
Impresión Clínica
Cuando se evaluó a la paciente en la unidad de alergia
e inmunología clínica, se obtuvieron más antecedentes. Además de la historia de
enteropatía autoinmune refractaria, había una historia de infección, que se
destacó por neumonía y otras infecciones sinopulmonares. Estas características,
en combinación con los antecedentes de dermatitis eccematosa, alergia
alimentaria, anemia hemolítica y proteinuria, sugerían un síndrome sistémico de
desregulación inmunitaria.
Se realizaron pruebas adicionales. Un hemograma
completo con recuento diferencial obtenido después de la interrupción de la
prednisona oral mostró eosinofilia. Los resultados de la electroforesis de
proteínas séricas y los niveles séricos de cadenas ligeras libres fueron
normales. Los niveles de IgG e IgM eran normales; Los niveles de IgE e IgA
estaban elevados. En las pruebas de anticuerpos funcionales, hubo niveles
protectores de tétanos IgG, Streptococcus pneumoniae IgG y Haemophilus
influenzaeIgG tipo b. La citometría de flujo mostró recuentos de células T
CD3+, CD4+ y CD8+ absolutos normales, junto con un recuento de células B CD19+
ligeramente bajo. El compartimiento de células T CD4+ estaba sesgado hacia un
fenotipo de memoria, y el compartimiento de CD8+ tenía una mayor expresión de
antígenos HLA de clase II que era consistente con un fenotipo activado. La
prueba de anticuerpos antienterocitos realizada en un laboratorio de referencia
fue positiva, con tinción lineal de IgG e IgA de las membranas apicales de los
enterocitos. Se sospechó un error innato de la inmunidad y se realizaron
pruebas genéticas.
DIAGNOSTICO CLÍNICO PRESUNTIVO
ERROR CONGÉNITO DE LA INMUNIDAD, MUY PROBABLEMENTE
DESREGULACIÓN INMUNE, POLIENDOCRINOPATÍA, ENTEROPATÍA, SÍNDROME LIGADO AL
CROMOSOMA X (IPEX).
Pruebas e Interpretación Genética
VARIANTE Y CLASIFICACIÓN
El paciente se sometió a pruebas genéticas basadas en paneles, que incluyeron secuenciación y análisis de deleción y duplicación de 207 genes asociados con inmunodeficiencias primarias y enfermedad inflamatoria intestinal monogénica. Esta prueba reveló tres variantes raras en tres genes diferentes: una variante sin sentido hemicigótica en FOXP3 , una variante promotora heterocigótica en el gen que codifica el componente de ARN de la endoribonucleasa procesadora de ARN mitocondrial ( RMRP ) y una variante donante de empalme heterocigótica en el gen que codifica Proteína 2 de unión al dominio SH3 ( SH3BP2 ) ( Tabla 2 ). El RMRP y SH3BP2No se pensó que las variantes fueran relevantes debido a la falta de coincidencia entre el fenotipo clínico del paciente y las asociaciones de enfermedades informadas y los patrones de herencia de los genes. Sin embargo, FOXP3 está asociado con el síndrome IPEX de enfermedad recesiva ligada al cromosoma X, por lo que la variante FOXP3 encontrada en este paciente justificaba una investigación adicional.

Tabla 2. Resultados de los test genéticos.
La variante FOXP3 fue clasificada por el laboratorio
de diagnóstico como una variante de significado incierto. Todas las variantes
detectadas mediante pruebas genéticas de diagnóstico clínico se clasifican de
acuerdo con las pautas publicadas por el Colegio Estadounidense de Genética
Médica y Genómica y la Asociación de Patología Molecular. 9Sobre la base de un
marco basado en la evidencia, a cada variante se le asigna una de cinco
clasificaciones: patógena, probablemente patógena, de significado incierto,
probablemente benigna o benigna, para ayudar a determinar la probabilidad de
que la variante cause la enfermedad. Las variantes de significado incierto son
hallazgos comunes en las pruebas genéticas que pueden ser difíciles de
interpretar. No pueden usarse solos para establecer un diagnóstico clínico o para
dirigir el manejo clínico. Los estudios de segregación familiar y los estudios
funcionales del producto génico pueden ayudar a determinar la relevancia
clínica de una variante de significado incierto.
Se realizaron estudios de segregación familiar para la
variante FOXP3 y mostraron que la variante se heredó de la madre del paciente,
que era portadora heterocigota. La variante estaba ausente en su padre y
hermano no afectado. Estos hallazgos son consistentes con el patrón de herencia
recesivo ligado al cromosoma X del síndrome IPEX.
MANIFESTACIONES DEL SÍNDROME IPEX Y EFECTOS SOBRE LAS
CÉLULAS T REGULADORAS
El síndrome IPEX es causado por variantes de pérdida
de función en FOXP3 , 10-12 , que es un regulador transcripcional maestro que
es fundamental para la función de las células T reguladoras CD4+ CD25+. 13 Las
células T reguladoras suprimen la activación y las funciones efectoras de los
linfocitos autorreactivos y potencialmente patógenos. 14 Las variantes de
pérdida de función en FOXP3 causan deficiencias cuantitativas o funcionales en
las células T reguladoras, lo que conduce a una desregulación inmunitaria. Esta
desregulación puede manifestarse como enfermedad autoinmune e inflamación
alérgica. 10,12,15,16
Además de la tríada clásica de manifestaciones
asociadas con el síndrome IPEX (endocrinopatías autoinmunes, enteropatía
autoinmune y dermatitis eczematosa), se han descrito muchas otras
manifestaciones, que incluyen anomalías neurológicas, trastornos hematológicos
(p. ej., citopenias inmunomediadas), enfermedad pulmonar, hepatitis ,
enfermedad renal e infecciones ( Figura 2 ). 17-20 La inflamación alérgica
puede incluir eccema, alergias alimentarias, eosinofilia y un nivel elevado de
IgE total. 20

Figura 2. Manifestaciones Clínicas del Síndrome IPEX.
La tríada clásica de manifestaciones asociadas con la
desregulación inmune, poliendocrinopatía, enteropatía, síndrome ligado al X
(IPEX)
comprende endocrinopatías autoinmunes, enteropatía
autoinmune y dermatitis eccematosa. Otras manifestaciones incluyen anomalías
neurológicas, trastornos hematológicos y enfermedades pulmonares,
gastrointestinales, renales e infecciosas. El sombreado rojo indica
manifestaciones que ocurrieron en este paciente.
Se han descrito variantes causantes de enfermedades en
las regiones codificantes y en las regiones no codificantes de FOXP3 ; no se
han identificado correlaciones claras de genotipo-fenotipo. 16,19-22 Cabe
destacar que las variantes de FOXP3 tienen una penetrancia variable, y la misma
variante causa diferentes presentaciones clínicas entre los portadores,
incluidos los miembros de la familia. 20
La presentación clínica de este paciente fue
consistente con el síndrome IPEX, y las pruebas genéticas mostraron una
variante de significado incierto en FOXP3 : FOXP3, exón 8, c.754A→G
(p.Lys252Glu). Este cambio de secuencia reemplazó la lisina con ácido glutámico
en el codón 252 de la proteína FOXP3. El residuo de lisina está moderadamente
conservado y existe una pequeña diferencia fisicoquímica entre la lisina y el
ácido glutámico. Esta mutación está en el dominio cremallera de leucina (
Figura 3 ), que media la homodimerización y la heterodimerización. 24 Esta
variante no está presente en las bases de datos de población (sin frecuencia en
la base de datos de agregación del genoma [gnomAD]). La predicción de
clasificación de intolerantes de tolerantes (SIFT) 25porque esta variante era
"perjudicial", y la predicción 26 de Polymorphism Phenotyping v2
(PolyPhen-2) era "probablemente dañina".

Figura 3. Dominios funcionales de FOXP3 y variante en
este paciente.
El síndrome IPEX es causado por variantes de pérdida
de función en el gen que codifica la proteína 3 de la caja de la cabeza de
horquilla (FOXP3). Estas variantes causan deficiencias cuantitativas o
funcionales en las células T reguladoras, lo que conduce a una desregulación
inmunitaria. Este la desregulación puede manifestarse como enfermedad
autoinmune e inflamación alérgica. Hay varios dominios funcionales del producto
génico. El dominio rico en prolina N-terminal regula la transcripción. El
dominio de dedo de zinc (ZF) está involucrado en las interacciones
proteína-ADN. El dominio cremallera de leucina (LZ) media la homodimerización y
la heterodimerización con FOXP3 y otros factores de transcripción de FOXP. El
dominio de unión al ADN de cabeza de horquilla (FKH) interactúa con el promotor
de interleucina-2 y otros genes diana. Este paciente tenía una variante en el
dominio LZ: c.754A→G (p.Lys252Glu). Esta variante no está presente en las bases
de datos de población (sin frecuencia en la base de datos de agregación del
genoma [gnomAD]). La predicción de clasificación de intolerantes de tolerantes
(SIFT) para esta variante fue "perjudicial" y el polimorfismo. La
predicción de Phenotyping v2 (PolyPhen-2) fue "probablemente dañina".
Secuencias potenciadoras no codificantes conservadas (SNC) son sitios diana de
modificaciones epigenéticas.23 E1 a E11 son los exones del transcrito canónico
FOXP3 (ID del transcrito Ensembl, ENST00000376207.4).
El siguiente paso fue realizar estudios funcionales
sobre las células T reguladoras del paciente. Los resultados del fenotipado de
las células T reguladoras, realizado en el Medical College of Wisconsin
mediante análisis de citometría de flujo con marcadores de superficie, fueron
anormales. Había un porcentaje total elevado de células T FOXP3+ CD4+ en
circulación, y estas células tenían una expresión atípica de CD25 y CTLA4. En
personas sanas, las células T reguladoras expresan constitutivamente altos
niveles de CD25 y CTLA4, porque FOXP3 impulsa su expresión. En este paciente,
muchas células T reguladoras FOXP3+ tenían expresión reducida de CTLA4 y CD25.
Dentro del espectro clínico del síndrome IPEX, se observa una disminución de
las células T reguladoras FOXP3+ circulantes en personas con variantes de FOXP3
que resultan en la pérdida de la expresión de proteínas. 17Por el contrario,
las personas con mutaciones sin sentido de FOXP3 pueden tener células T
reguladoras en las que FOXP3 está presente pero funciona parcialmente o no
funciona. 16,18 Este paciente tenía una variante sin sentido en la que estaba
presente FOXP3, pero parece haber tenido una variante hipomórfica que afectaba
la función de FOXP3. En general, los resultados de la citometría de flujo en
este paciente fueron consistentes con la disfunción de las células T
reguladoras y la hiperactivación de las células T, hallazgos que sugieren una
variante hipomórfica de FOXP3 .
En este paciente, la historia clínica, la detección de
una variante FOXP3 que también estaba presente en su madre (hallazgo compatible
con herencia ligada al cromosoma X), los resultados anormales en el fenotipado
de células T reguladoras mediante análisis de citometría de flujo con
marcadores de superficie , y la demostración de un aumento en las células T
CD4+ y CD8+ efectoras y activadas en circulación llevó al diagnóstico de
síndrome IPEX.
Diagnóstico Genético
Desregulación inmune, poliendocrinopatía, enteropatía,
síndrome ligado al X (IPEX) causado por una variante hemicigota en FOXP3 .
DISCUSIÓN DE MANEJO
Mientras estaban pendientes los resultados de más
pruebas de diagnóstico, el enfoque del tratamiento se centró en el control de
los síntomas. Inicialmente se continuó con prednisona y budesonida. Un ensayo
que involucró la interrupción de losartán no mejoró la condición del paciente,
por lo que se reinició la terapia con losartán para el tratamiento de la
hipertensión. Los síntomas no remitieron y se inició tratamiento con
vedolizumab intravenoso. Vedolizumab es un bloqueador de la integrina α 4 β 7
que funciona uniéndose al receptor y previniendo la interacción con la molécula
de adhesión celular 1 de la dirección de la mucosa.
Después de 3 meses de tratamiento, la frecuencia de
las deposiciones disminuyó a una vez cada dos días y la dosis de prednisona se
redujo con éxito. Sin embargo, después de 6 meses de tratamiento, los síntomas
recurrieron, con solo una mejoría transitoria después de intensificar la dosis
de vedolizumab. Se inició nuevamente tratamiento con abatacept intravenoso, con
mejoría inicial; posteriormente continuó la diarrea frecuente con
deshidratación y empeoró la proteinuria. Se discutió con el paciente una opción
alternativa para una terapia más definitiva, y optó por una remisión para ser
evaluado para un trasplante alogénico de células hematopoyéticas (HCT).
El HCT se usa cada vez más para el tratamiento de
enfermedades no malignas, incluidos los síndromes de insuficiencia de la médula
ósea, las hemoglobinopatías, los errores innatos de la inmunidad y los
trastornos metabólicos hereditarios. La justificación para el uso de HCT en el
tratamiento de pacientes con síndrome IPEX es sencilla: la restauración de las
células T reguladoras funcionales debería restaurar el equilibrio inmunológico
normal, brindando alivio de las manifestaciones autoinmunes y obviando la
necesidad de una terapia inmunosupresora continua. Múltiples informes de casos
y pequeñas series han mostrado éxito con el uso de HCT en pacientes con
síndrome IPEX. También se informó una gran serie internacional que involucró a
96 pacientes con síndrome IPEX que fueron tratados en 38 instituciones. 20Los
pacientes tratados solo con terapia médica tenían manifestaciones de por vida y
necesitaban terapia inmunosupresora continua. Entre los 58 pacientes que se
sometieron a TCH, la supervivencia global fue del 73,2 % con una mediana de
seguimiento de 2,7 años. La extensión y la gravedad de la afectación de órganos
antes del TCH predijeron los resultados después del TCH.
Dada la ausencia de condiciones coexistentes y la
enfermedad bien controlada en este paciente, se recomendó HCT. Una búsqueda
exhaustiva de donantes reveló que su hermano era haploidéntico (y negativo para
el FOXP3 familiar).variante) y también identificó varios donantes no
emparentados totalmente compatibles. En el momento en que se estaba
planificando el TCH, la pandemia de la enfermedad por coronavirus 2019
(Covid-19) dificultó bastante la obtención de médula ósea fresca de donantes no
emparentados, por lo que se tomó la decisión de proceder con el trasplante de
médula ósea haploidéntica con su hermano como donante. Seguimos nuestra plataforma
estándar de HCT de intensidad reducida que usamos para enfermedades no
malignas, que implica el uso de globulina anti-células T, una fuente de injerto
de médula ósea y dosis altas de ciclofosfamida para la prevención de la
enfermedad de injerto contra huésped, como así como la reducción gradual tardía
de la terapia inmunosupresora. 27,28
El paciente tuvo las complicaciones tempranas
esperadas asociadas con el HCT haploidéntico, pero se recuperó con un injerto
enérgico derivado del donante. Dos meses después del TCH, recibió un
diagnóstico de cistitis hemorrágica asociada al virus BK. Tres meses después
del TCH, la diarrea reapareció y los estudios de quimerismo mostraron un
rechazo secundario del injerto con recuperación hematopoyética autóloga. Se añadió
prednisona para controlar la diarrea, y posteriormente se admitió al paciente
para un segundo TCH alogénico, esta vez con acondicionamiento de mayor
intensidad, sin el uso de globulina anti-células T y con el uso de médula ósea
fresca de un paciente completamente nuevo. donante no emparentado compatible.
No hubo complicaciones clínicamente significativas después del segundo HCT, y
el injerto de neutrófilos del donante se estableció el día 16 después del
trasplante. Ocho meses después del segundo HCT, el paciente no ha tenido
enfermedad de injerto contra huésped y tiene un funcionamiento gastrointestinal
normal. La dosis de inmunosupresión sistémica se está reduciendo y
recientemente regresó al trabajo con un estado funcional completo.
DIAGNOSTICO FINAL
DESREGULACIÓN INMUNE, POLIENDOCRINOPATÍA, ENTEROPATÍA,
SÍNDROME LIGADO AL CROMOSOMA X (IPEX).
Traducción de:
Francis P. Colizzo, M.D., Stuti G. Shroff, M.B., B.S.,
Ph.D., Frances A. High, M.D., Ph.D., Yi-Bin Chen, M.D., and Sara Barmettler,
M.D.
https://www.nejm.org/doi/full/10.1056/NEJMcpc2201236
1. Gentile NM, Murray JA,
Pardi DS.
Autoimmune enteropathy: a
review and
update of clinical management.
Curr Gas[1]troenterol
Rep 2012;14:380-5.
2. Rubio-Tapia A, Hill
ID, Kelly CP,
Calderwood AH, Murray JA.
ACG clinical
guidelines: diagnosis and
management of
celiac disease. Am J
Gastroenterol 2013;
108:656-77.
3. Moran CJ, Klein C,
Muise AM, Snap[1]per
SB. Very early-onset inflammatory
bowel disease: gaining
insight through
focused discovery.
Inflamm Bowel Dis
2015;21:1166-75.
4. Seidelin JB, Riis LB,
Butt RA. Crohn’s
disease with progressive
renal impair[1]ment.
Gastroenterology 2020;158:58-9.
5. Ambruzs JM, Walker PD,
Larsen CP.
The histopathologic
spectrum of kidney
biopsies in patients with
inflammatory
bowel disease. Clin J Am
Soc Nephrol
2014;9:265-70.
6. Kamal A, Fain C, Park
A, et al. Angio[1]tensin
II receptor blockers and gastroin[1]testinal
adverse events of resembling
sprue-like enteropathy: a
systematic review.
Gastroenterol Rep (Oxf)
2019;7:162-7.
7. Kluger N, Jokinen M,
Krohn K, Ranki
A. Gastrointestinal
manifestations in
APECED syndrome. J Clin
Gastroenterol
2013;47:112-20.
8. Braskett MJ, Chatila
T. IPEX: immune
dysregulation,
polyendocrinopathy, enter[1]opathy,
X-linked. UpToDate. 2022 (https://
www.uptodate.com/contents/ipex-immune
-dysregulation-polyendocrinopathy
-enteropathy-x-linked).
9. Richards S, Aziz N,
Bale S, et al. Stan[1]dards
and guidelines for the interpreta[1]tion
of sequence variants: a joint consen[1]sus
recommendation of the American
College of Medical
Genetics and Genom[1]ics
and the Association for Molecular Pa[1]thology.
Genet Med 2015;17:405-24.
10. Chatila TA, Blaeser
F, Ho N, et al.
JM2, encoding a fork
head-related protein,
is mutated in X-linked
autoimmunity[1]allergic
disregulation syndrome. J Clin
Invest
2000;106(12):R75-R81.
11. Wildin RS, Ramsdell
F, Peake J, et al.
X-linked neonatal
diabetes mellitus, en[1]teropathy
and endocrinopathy syndrome
is the human equivalent
of mouse scurfy.
Nat Genet 2001;27:18-20.
12. Bennett CL, Christie
J, Ramsdell F, et al.
The immune dysregulation,
polyendocri[1]nopathy,
enteropathy, X-linked syndrome
(IPEX) is caused by
mutations of FOXP3.
Nat Genet 2001;27:20-1.
13. Zheng Y, Rudensky AY.
Foxp3 in con[1]trol
of the regulatory T cell lineage. Nat
Immunol 2007;8:457-62.
14. Sakaguchi S,
Yamaguchi T, Nomura
T, Ono M. Regulatory T
cells and immune
tolerance. Cell
2008;133:775-87.
15. Gavin MA, Torgerson
TR, Houston E,
et al. Single-cell
analysis of normal and
FOXP3-mutant human T
cells: FOXP3 ex[1]pression
without regulatory T cell devel[1]opment.
Proc Natl Acad Sci U S A 2006;
103:6659-64.
16. d’Hennezel E,
Ben-Shoshan M, Ochs
HD, et al. FOXP3 forkhead
domain muta[1]tion
and regulatory T cells in the IPEX
syndrome. N Engl J Med
2009;361:1710-3.
17. Torgerson TR, Ochs
HD. Immune
dysregulation,
polyendocrinopathy, enter[1]opathy,
X-linked: forkhead box protein 3
mutations and lack of
regulatory T cells.
J Allergy Clin Immunol
2007;120:744-52.
18. Verbsky JW, Chatila
TA. Immune dys[1]regulation,
polyendocrinopathy, enterop[1]athy,
X-linked (IPEX) and IPEX-related
disorders: an evolving
web of heritable
autoimmune diseases. Curr
Opin Pediatr
2013;25:708-14.
19. Gambineri E, Perroni
L, Passerini L,
et al. Clinical and
molecular profile of a
new series of patients
with immune dys[1]regulation,
polyendocrinopathy, enterop[1]athy,
X-linked syndrome: inconsistent cor[1]relation
between forkhead box protein 3
expression and disease
severity. J Allergy
Clin Immunol
2008;122(6):1105-1112.e1.
20. Barzaghi F, Amaya
Hernandez LC,
Neven B, et al. Long-term
follow-up of IPEX
syndrome patients after
different thera[1]peutic
strategies: an international multi[1]center
retrospective study. J Allergy Clin
Immunol
2018;141(3):1036-1049.e5.
21. Wildin RS,
Smyk-Pearson S, Filipov[1]ich
AH. Clinical and molecular features of
the immunodysregulation,
polyendocri[1]nopathy,
enteropathy, X linked (IPEX)
syndrome. J Med Genet
2002;39:537-45.
22. Barzaghi F, Passerini
L, Bacchetta R.
Immune dysregulation,
polyendocrinop[1]athy,
enteropathy, x-linked syndrome:
a paradigm of
immunodeficiency with
autoimmunity. Front
Immunol 2012;3:
211.
23. Bacchetta R, Barzaghi
F, Roncarolo
M-G. From IPEX syndrome
to FOXP3 mu[1]tation:
a lesson on immune dysregula[1]tion.
Ann N Y Acad Sci 2018;1417:5-22.
24. Chae W-J, Henegariu
O, Lee S-K,
Bothwell ALM. The mutant
leucine-zipper
domain impairs both
dimerization and
suppressive function of
Foxp3 in T cells.
Proc Natl Acad Sci U S A
2006;103:9631-6.
25. Sim NL, Kumar P, Hu
J, Henikoff S,
Schneider G, Ng PC. SIFT
web server: pre[1]dicting
effects of amino acid substitu[1]tions
on proteins. Nucleic Acids Res 2012;
40:W452-W457.
26. Adzhubei I, Jordan
DM, Sunyaev SR.
Predicting functional
effect of human
missense mutations using
PolyPhen-2.
Curr Protoc Hum Genet
2013;7:720.
27. DeZern AE, Zahurak
ML, Symons HJ,
et al. Haploidentical BMT
for severe aplas[1]tic
anemia with intensive GVHD prophy[1]laxis
including posttransplant cyclophos[1]phamide.
Blood Adv 2020;4:1770-9.
28. Leick M, Hunter B,
DeFilipp Z, et al.
Posttransplant
cyclophosphamide in allo[1]geneic
bone marrow transplantation for
the treatment of
nonmalignant hemato[1]logical
diseases. Bone Marrow Transplant
2020;55:758-62.