Una mujer de 41 años con disartria transitoria y caída facial fue trasladada a este hospital para evaluación de masas en la válvula mitral.
Tres semanas antes de esta presentación, la paciente
acudió al departamento de emergencias de otro hospital debido a preocupaciones
sobre el sangrado vaginal abundante que se había desarrollado 1 semana antes y
se asoció con dolor abdominal tipo cólico, fatiga, disnea y mareos. En el
examen se destacó una frecuencia cardíaca de 91 latidos por minuto, una presión
arterial de 130/79 mm Hg y una saturación de oxígeno del 100% mientras
respiraba aire ambiente; por lo demás, según los informes, el examen fue
normal, aunque no se realizó un examen ginecológico. Los resultados de
laboratorio fueron notables por un nivel de hemoglobina de 8,2 g por decilitro
(rango de referencia, 12,0 a 16,0; nivel previo 1 año antes, 9,1), un volumen
corpuscular medio de 71,3 fl (rango de referencia, 80 a 94) y un recuento de
plaquetas de 148.000 por microlitro (rango de referencia, 150.000 a 450.000).
La gonadotropina coriónica humana en orina fue indetectable. Se administró solución
salina normal por vía intravenosa y se prescribió un ciclo de acetato de
medroxiprogesterona durante 5 días.
Once días después de que la paciente completó la
terapia con acetato de medroxiprogesterona (9 días antes de esta presentación),
recurrió el sangrado vaginal abundante y la disnea. Se prescribió un curso
adicional de 3 días de acetato de medroxiprogesterona. La noche después de que
la paciente terminó el segundo curso de tratamiento, dentro de 1 hora después
de haberla visto y parecía estar bien, su esposo notó que tenía caída facial en
el lado derecho y dificultad para hablar. Llamó a los servicios médicos de emergencia.
Cuando llegó al servicio de urgencias de un segundo hospital, dentro de 1 hora
después del inicio de los síntomas, en su examen destacaba una frecuencia
cardíaca de 91 latidos por minuto, una presión arterial de 173/83 mm Hg y una
saturación de oxígeno de 99% mientras respiraba aire ambiente. Se observó que
tenía caída facial en el lado derecho y habla ininteligible.
La tomografía computarizada (TC) de la cabeza ( Figura
1A ) no reveló evidencia de infarto agudo o hemorragia intracraneal. Se
observaron pequeños infartos crónicos en el tálamo derecho y el hemisferio
cerebeloso derecho. Un angiograma por TC (CTA) de la cabeza y el cuello no
mostró estenosis de alto grado ni oclusión de grandes vasos.

Figura 1. Estudios de Neuroimagen.
Una imagen de tomografía computarizada axial inicial
de la cabeza, realizada sin la administración de material de contraste
intravenoso (Panel A), no muestra anormalidad intracraneal. No hubo infarto
grande ni hemorragia intracraneal. Al día siguiente, se realizó resonancia
magnética de la cabeza. Las imágenes axiales ponderadas en T2 muestran infartos
crónicos en el tálamo derecho (Panel B, flecha) y el hemisferio cerebeloso
derecho (Panel C, flecha). Una imagen ponderada por difusión axial (Panel D) no
muestra áreas de difusión restringida que sugieran una enfermedad aguda. Las
imágenes de proyección de máxima intensidad se obtuvieron a partir de angiografía
por resonancia magnética tridimensional (ARM), en tiempo de vuelo de la
circulación intracraneal (Panel E) y ARM del cuello realzada con gadolinio
(Panel F). Las imágenes no muestran estenosis de alto grado u oclusión de las
principales arterias intracraneales o cervicales.
Se obtuvo una consulta de neurología; Se administró
aspirina y atorvastatina. Los niveles sanguíneos de electrolitos, albúmina,
colesterol de lipoproteínas de baja densidad y hemoglobina glicosilada eran
normales, al igual que el recuento de glóbulos blancos y el recuento
diferencial y los resultados de las pruebas de función hepática y renal. La
gonadotropina coriónica humana en orina fue indetectable y el análisis de orina
mostró sangre 3+, sin proteínas ni cilindros. En la Tabla 1 se muestran
resultados de pruebas de laboratorio adicionales . Dentro de los 45 minutos
posteriores a la llegada del paciente al departamento de emergencias del
segundo hospital, la asimetría facial y la disartria se resolvieron. La
paciente fue ingresada en la unidad médica del segundo hospital.

Tabla 1. Datos de laboratorio.
Al día siguiente, la resonancia magnética nuclear
(RMN) y la angiografía de la cabeza y el cuello revelaron nuevamente infartos
lacunares antiguos en el tálamo derecho ( Figura 1B ) y el hemisferio
cerebeloso derecho ( Figura 1C ). No hubo evidencia de infarto agudo ( Figura
1D ), hemorragia intracraneal u oclusión cerebrovascular ( Figura 1E y 1F ).
Se transfundieron dos unidades de concentrados de
glóbulos rojos. Los resultados de las pruebas de laboratorio posteriores a la
transfusión se muestran en la Tabla 1 . Un ecocardiograma transtorácico (
Figura 2A y 2B ) mostró valvas de la válvula mitral engrosadas e insuficiencia
mitral de leve a moderada, con una presión sistólica del ventrículo derecho
estimada en 48 mm Hg .
En el tercer día de hospitalización, un ecocardiograma
transesofágico ( Figura 2C ) mostró ecodensidades múltiples en el lado
auricular de las valvas de la válvula mitral en las líneas de cierre que eran
consistentes con vegetaciones. Se obtuvieron cultivos de sangre y se administró
heparina por vía intravenosa.
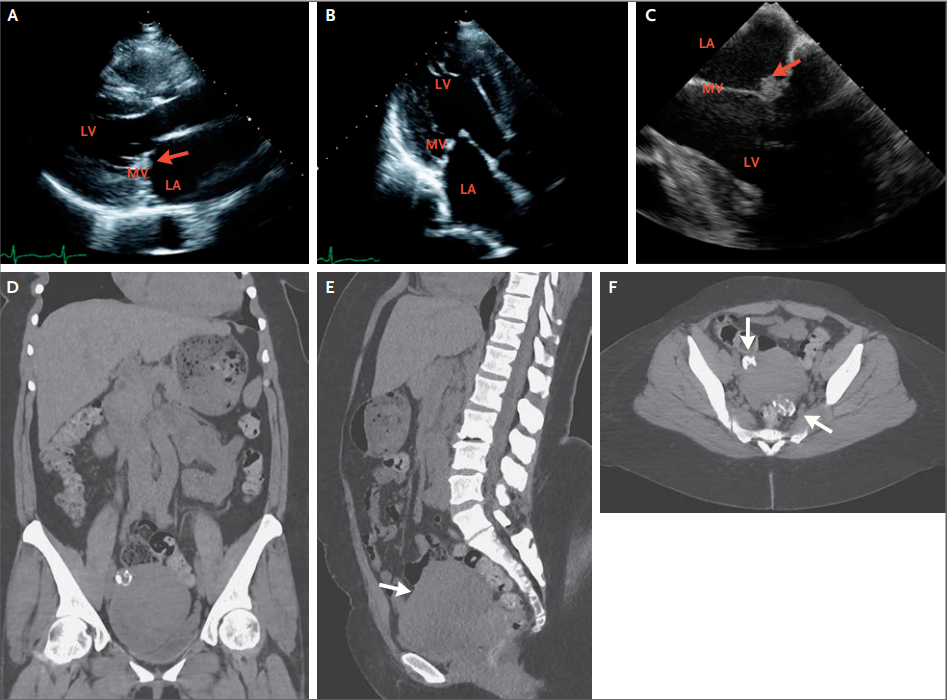
Figura 2.Ecocardiogramas y TAC de Abdomen y Pelvis.
En la ecocardiografía transtorácica (Panel A), una
vista de eje largo paraesternal muestra engrosamiento en las puntas de las
valvas de la válvula mitral, un hallazgo sugestivo de vegetaciones (flecha).
Una vista apical de tres cámaras en la ecocardiografía transtorácica (Panel B)
nuevamente muestra engrosamiento en las puntas de las valvas de la válvula
mitral en la superficie auricular. Un ecocardiograma transesofágico (Panel C)
muestra las vegetaciones (flecha) en el lado atrial de las valvas de la válvula
mitral en las líneas de cierre. Hubo insuficiencia mitral leve a moderada asociada
(no se muestra). Las imágenes de TC coronal (Panel D) y sagital (Panel E) del
abdomen y la pelvis, obtenidas sin la administración de material de contraste
intravenoso, muestran un útero heterogéneo agrandado (Panel E, flecha). Una
imagen de TC axial de la pelvis (Panel F) muestra masas uterinas calcificadas
(flechas) que eran consistentes con fibromas. LA denota aurícula izquierda,
ventrículo izquierdo LV y válvula mitral MV.
En el cuarto día de hospitalización, la tasa de
infusión de heparina se redujo en intervalos de 6 horas cuando el tiempo de
tromboplastina parcial excedía los 150 segundos, hasta que se detuvo la
infusión. Los resultados de las pruebas de laboratorio se muestran en la Tabla
1 ; los niveles de complemento (C3 y C4) eran normales. Se obtuvieron cultivos
de sangre adicionales.
Una nueva tomografía computarizada de la cabeza no
reveló anomalías agudas. La TC de tórax mostró nódulos agrupados en los lóbulos
inferiores que probablemente estaban relacionados con aspiración, y la TC de abdomen
y pelvis ( Figura 2D, 2E y 2F ) reveló un útero heterogéneo agrandado con
múltiples fibromas.
Al día siguiente, la paciente fue trasladada a este
hospital. En una revisión de los sistemas, no informó recurrencia de disartria
o caída facial y no notó otros síntomas neurológicos. Informó que había tenido
sangrado menstrual abundante y disnea de esfuerzo en las semanas anteriores a
esta presentación; erupciones maculares pruriginosas en el tronco, el cuero
cabelludo, los brazos y las piernas con fotosensibilidad asociada que había
persistido durante muchas semanas; nuevo fenómeno de Raynaud en los últimos 10
meses; y una larga historia de ojos secos y alergias estacionales. El historial
médico fue notable por el rasgo de células falciformes, deficiencia de vitamina
B 12, deficiencia de hierro que resultó en infusiones de hierro en el pasado,
fibromas uterinos, ansiedad e hipertensión. Los antecedentes quirúrgicos
incluían múltiples laparoscopias y dos laparotomías, incluida
salpingo-ooforectomía por endometriosis. Debido a la infertilidad, se había
sometido a una fecundación in vitro; tuvo un aborto espontáneo en el primer
trimestre a los 25 años de edad. Durante su segundo embarazo a los 33 años de
edad, una prueba de detección de reagina plasmática rápida (RPR) fue positiva
en 1:8; sin embargo, una prueba confirmatoria treponémica específica para
sífilis fue negativa. Se desarrolló hipertensión inducida por el embarazo,
seguida de desprendimiento de la placenta a las 30 semanas, lo que resultó en
un parto por cesárea. Los medicamentos incluyeron amlodipina, cianocobalamina,
sulfato ferroso y acetato de medroxiprogesterona. No tenía alergias conocidas a
medicamentos.
La paciente vivía en el este de Massachusetts con su
esposo y su hijo. No bebía alcohol ni fumaba tabaco. Su madre tenía artritis
reumatoide, hipertensión y antecedentes de aborto espontáneo. Una abuela tenía
diabetes y la otra abuela había tenido un stroke. Una tía había tenido leucemia
y luego cáncer de mama. Su hijo tiene psoriasis.
La temperatura era de 36,8°C, la frecuencia cardíaca
de 74 latidos por minuto, la presión arterial de 155/98 mm Hg y la saturación
de oxígeno del 99% mientras el paciente respiraba aire ambiente. La altura era
de 175 cm, el peso de 107 kg y el índice de masa corporal 34,9. Tenía varias
máculas circulares pequeñas en la espalda ( Figura 3 ). Soplo holosistólico
grado 2/6 en borde y ápex esternal izquierdo. No tenía linfadenopatía,
petequias, cambios en las uñas o evidencia de artritis. El resto del examen fue
normal, al igual que un examen neurológico.

Figura 3. Fotografía de las Máculas en la Espalda.
En la presentación en este hospital, varios pequeñas
máculas circulares hiperpigmentadas, que la paciente describió como
pruriginosas y fotosensibles, se observaron en la espalda.
Los resultados de las pruebas de laboratorio se
muestran en la Tabla 1 . Todos los hemocultivos obtenidos en el segundo
hospital no mostraron crecimiento. Un electrocardiograma fue normal.
Se recibieron los resultados de las pruebas de diagnóstico
y se tomaron las decisiones de manejo.
DIAGNÓSTICO DIFERENCIAL
El primer paso para desarrollar un diagnóstico
diferencial en este caso es determinar si la presentación de este paciente está
relacionada con una afección inflamatoria o no inflamatoria. Los trastornos no
inflamatorios a considerar incluyen ataque isquémico transitorio (AIT) o
accidente cerebrovascular debido a enfermedad aterosclerótica o embolia,
endocarditis infecciosa con cultivo negativo, sangrado inducido por fibroma y
cáncer. Las posibles causas inflamatorias incluyen endocarditis trombótica no
bacteriana, síndrome antifosfolípido (SAF), lupus eritematoso sistémico (LES) y
vasculitis. Me concentraré en las características individuales de la
presentación de esta paciente y veré si hay alguna pista que apunte a uno o más
de estos diagnósticos.
TIEMPO PROLONGADO DE TROMBOPLASTINA PARCIAL Y SANGRADO
Una característica notable de la presentación de esta
paciente es el sangrado vaginal en el contexto de un tiempo de tromboplastina
parcial levemente prolongado. Esto puede deberse a la anticoagulación, a una
deficiencia de factor intrínseco (como la que ocurre en pacientes con
hemofilia) o a la presencia de un inhibidor de la coagulación. Los inhibidores
adquiridos pueden desarrollarse durante el embarazo o en asociación con cáncer
o enfermedades del tejido conjuntivo, pero también pueden observarse en
personas sanas no embarazadas. Además, la prolongación del tiempo de
tromboplastina parcial puede indicar la presencia de anticoagulante lúpico, que
generalmente se determina mediante pruebas secuenciales. En primer lugar, se
realiza una prueba de detección dependiente de fosfolípidos, como la prueba de
tiempo de veneno de víbora de Russell diluido o una prueba de tiempo de tromboplastina
parcial activada (aPTT, por sus siglas en inglés) sensible al anticoagulante
lúpico. Después, el fracaso de los estudios de mezcla para corregir la
prolongación del aPTT indica la presencia de un inhibidor, en lugar de una
deficiencia de factor. Finalmente, si la adición de exceso de fosfolípidos
corrige la prolongación, se confirma la presencia de anticoagulante lúpico. A
pesar del nombre, el anticoagulante lúpico generalmente provoca la coagulación
en el sistema venoso o arterial y rara vez provoca sangrado. Por lo tanto, el
sangrado vaginal en esta paciente puede ser causado por otro trastorno como los
fibromas uterinos.
RESULTADO FALSO POSITIVO EN LA PRUEBA RÁPIDA DE
REAGINA PLASMÁTICA
Los resultados falsos positivos en pruebas serológicas
no treponémicas para sífilis generalmente son de títulos bajos. Es importante
confirmar cualquier prueba no treponémica reactiva, como la prueba RPR, con una
prueba treponémica específica. Dado que la prueba RPR de este paciente no
confirmó un diagnóstico de sífilis, es importante comprender las condiciones
asociadas con un resultado RPR falso positivo. Puede producirse un resultado
falso positivo debido a la detección de anticuerpos anticardiolipina, ya que el
antígeno de la prueba RPR contiene cardiolipina. Un resultado RPR falso
positivo también puede ocurrir en el contexto del embarazo, enfermedades
febriles agudas (p. ej., endocarditis), enfermedad hepática crónica, infección
por el virus de la inmunodeficiencia humana, inmunizaciones recientes, uso de
drogas por vía intravenosa y enfermedades autoinmunes como SAF y LES.
ATAQUE ISQUÉMICO TRANSITORIO E HISTORIAL DE
COMPLICACIONES DEL EMBARAZO
La causa más probable de AIT en esta paciente son los
fenómenos embólicos de la vegetación valvular. Aunque las mujeres que han
tenido un embarazo complicado anterior tienen un mayor riesgo de accidente
cerebrovascular que las mujeres con embarazos previos sin complicaciones, 1la
combinación de complicaciones del embarazo, aborto espontáneo y fenómenos
tromboembólicos en esta paciente despierta la sospecha de SAF. El SAF es un
síndrome autoinmune caracterizado por trombosis venosa o arterial,
complicaciones del embarazo, o ambas, en el contexto de una prueba de
anticuerpos antifosfolípidos positiva. El SAF puede ser un trastorno primario o
puede ser una consecuencia del LES u otra enfermedad autoinmune. Los criterios
revisados de Sapporo, también conocidos como criterios de Sydney, pueden ser
útiles en el diagnóstico de SAF, que requiere que un paciente cumpla con un
criterio clínico y un criterio de laboratorio. 2 Los criterios clínicos
incluyen trombosis vascular o complicaciones del embarazo, y los criterios de
laboratorio requieren la presencia de uno o más anticuerpos antifosfolípidos
con pruebas de confirmación. 2Aunque aún no sabemos si esta paciente tiene
anticuerpos antifosfolípidos, hubo preocupación por la presencia de dichos
anticuerpos, dada la historia de una prueba de RPR positiva y un tiempo de
tromboplastina parcial prolongado.
ERUPCIÓN FOTOSENSIBLE
La naturaleza fotosensible de la erupción de esta
paciente aumenta la sospecha de enfermedad del tejido conjuntivo, en particular
lupus eritematoso cutáneo subagudo, quizás la forma papuloescamosa, ya que la
apariencia anular típica no es evidente. Una biopsia de la erupción puede ser
útil, aunque no siempre es necesaria; una muestra de piel puede mostrar dermatitis
de interfase vacuolar, que puede ocurrir con la enfermedad del tejido
conectivo. El lupus eritematoso cutáneo subagudo a menudo se asocia con la
presencia de anticuerpos anti-Ro (SSA). La erupción que se observa en el cuero
cabelludo de esta paciente genera preocupación por el lupus discoide, que
generalmente afecta la cara, el cuero cabelludo, las orejas y el cuello y puede
estar asociado con taponamiento folicular y cicatrización. Estas erupciones en
combinación con el fenómeno de Raynaud de nueva aparición pueden estar
asociadas con una enfermedad del tejido conectivo como el LES.
ENDOCARDITIS TROMBÓTICA NO BACTERIANA
Las vegetaciones valvulares identificadas en imágenes
cardíacas en el contexto de hemocultivos negativos y sin signos claros de
infección sugieren endocarditis trombótica no bacteriana. Esta condición se
caracteriza por trombos de plaquetas estériles que comúnmente se adhieren a las
válvulas mitral y aórtica. Esto es distinto de la endocarditis bacteriana con
cultivo negativo, que es causada por microorganismos que son difíciles de
cultivar. La endocarditis trombótica no bacteriana a menudo se asocia con cáncer
avanzado, pero también puede ocurrir en pacientes con LES o SAF. La
endocarditis trombótica no bacteriana se conoce comúnmente como endocarditis de
Libman-Sacks en el contexto de LES o SAF, una entidad descrita por primera vez
en 1924 por Emanuel Libman y Benjamin Sacks. 3 Hasta el 50% de los pacientes
con LES o SAF tienen anomalías valvulares. 4
ESTABLECIENDO UN DIAGNÓSTICO
La constelación de AIT de esta paciente, infartos
crónicos en las imágenes, complicaciones del embarazo, un tiempo de
tromboplastina parcial prolongado y un resultado falso positivo en la prueba RPR
es altamente sugestivo de SAF. Dado el informe de una erupción fotosensible y
el fenómeno de Raynaud de nueva aparición, hubo preocupación por el diagnóstico
de LES. Finalmente, las vegetaciones no bacterianas de la válvula mitral y el
engrosamiento de la valva de la válvula mitral en el contexto de todos estos
hallazgos son más consistentes con endocarditis de Libman-Sacks. Juntando todas
las características de este caso, lo más probable es que este paciente tenga
endocarditis de Libman-Sacks con SAF y LES asociados.
IMPRESIÓN CLÍNICA
El examen de esta paciente reveló dos formas
morfológicas de erupción. Máculas eritematosas e hiperpigmentadas
fotodistribuidas con escamas ligeras estaban presentes en el pecho, la espalda
y la parte superior de los brazos y sugerían lupus eritematoso cutáneo
subagudo. Se observó alopecia cicatricial en las regiones occipital, nucal,
vértice izquierdo y parietal derecho del cuero cabelludo con hiperqueratosis
asociada y áreas de hiperpigmentación e hipopigmentación. La paciente informó
una erupción anterior similar que involucraba los pabellones auriculares. La
alopecia cicatricial y los hallazgos asociados eran indicativos de lupus
discoide. Se aplazó una biopsia de piel. La combinación de lupus discoide,
lupus eritematoso cutáneo subagudo, endocarditis trombótica no bacteriana y los
hallazgos de laboratorio que se describen a continuación sugieren el
diagnóstico clínico de LES. Aunque los niveles sanguíneos de C3 y C4 eran
normales, otros fenómenos inmunológicos apoyan el diagnóstico de LES.
DIAGNOSTICO CLÍNICO PRESUNTIVO
ENDOCARDITIS DE LIBMAN-SACKS CON SÍNDROME
ANTIFOSFOLÍPIDO ASOCIADO Y LUPUS ERITEMATOSO SISTÉMICO.
DISCUSIÓN PATOLÓGICA
Las pruebas de laboratorio iniciales revelaron un
tiempo de tromboplastina parcial levemente prolongado con un tiempo de
protrombina normal. Las causas comunes de tiempo de tromboplastina parcial
prolongado aislado incluyen la administración de heparina, ciertas deficiencias
de factores de coagulación y la presencia de anticoagulante lúpico. No se
detectó heparina en la muestra de sangre de este paciente, y los resultados de
los estudios del factor de coagulación en el tiempo con tromboplastina parcial
fueron normales ( Tabla 2). Una prueba de detección de aPTT sensible al
anticoagulante lúpico fue positiva y la muestra de sangre no corrigió la
prolongación del aPTT cuando se mezcló con plasma normal. La presencia de
anticoagulante lúpico se confirmó cuando el tiempo de tromboplastina parcial se
acortó después de la adición de un exceso de fosfolípido. Pruebas adicionales
revelaron niveles elevados de anticardiolipina IgG y anti-β 2 -glucoproteína 1
IgG. Tomados en conjunto, estos resultados constituyen hallazgos
"triplemente positivos" para la presencia de anticoagulante lúpico,
la explicación más probable para el tiempo prolongado de tromboplastina parcial
de este paciente. Para cumplir con los criterios de laboratorio para SAF, una
prueba de anticoagulante lúpico o una prueba para cualquiera de los anticuerpos
repetida al menos 12 semanas después debe mostrar un resultado positivo. 6

Tabla 2. Datos de laboratorio adicionales.
Algunas pruebas de laboratorio que se realizaron en el
segundo hospital se repitieron al presentarse en este hospital, lo que resultó
en conjuntos duplicados de resultados ( Tabla 2 ). Un ensayo en el que se
utilizó sustrato de células HEp-2 para confirmar la presencia de anticuerpos
antinucleares en una muestra evaluada en el laboratorio de nuestro hospital
resultó positivo a un título de 1:160 con un patrón homogéneo. Una prueba de
anticuerpos antinucleares que se había realizado en el segundo hospital 5 días
antes fue positiva a un título de 1:320 con un patrón similar ( Tabla 2). Las
pruebas de anticuerpos anti-Smith y anti-RNP fueron débilmente positivas cuando
se realizaron en el segundo hospital; los resultados fueron normales en nuestro
laboratorio. Los anticuerpos anti-Ro (SSA) estuvieron presentes en las muestras
evaluadas en ambos laboratorios. Los resultados de las pruebas de anticuerpos
anti-La (SSB) y anticuerpos anti-ADN de doble cadena fueron normales en ambos
laboratorios.
Los resultados obtenidos en el laboratorio del segundo
hospital fueron similares pero no idénticos a los obtenidos en nuestro
laboratorio; esto puede explicarse por la variación biológica a lo largo del
tiempo, factores preanalíticos como la manipulación de muestras o la
variabilidad entre ensayos. Las pruebas de competencia a nivel nacional, en las
que se envían muestras idénticas a cientos de laboratorios para su comparación,
muestran un grado similar de variabilidad entre plataformas. 7 Esta variabilidad
agrava la dificultad de desarrollar criterios de laboratorio estrictos para el
LES. 8 Aunque el diagnóstico de LES no depende de un resultado de laboratorio,
9la combinación de pruebas positivas de anticuerpos antinucleares, pruebas
positivas de anticoagulante lúpico y la presencia de anticuerpos anti-Smith en
este paciente respalda el diagnóstico clínico de LES.
DIAGNÓSTICO DE LABORATORIO
COMPATIBLE CON LUPUS ERITEMATOSO SISTÉMICO Y SÍNDROME
ANTIFOSFOLÍPIDO.
DISCUSIÓN DEL MANEJO
La hidroxicloroquina se inició para el tratamiento del
LES con lupus cutáneo activo, junto con un énfasis en la protección de la piel
contra la luz ultravioleta. Con raras excepciones, la hidroxicloroquina está
indicada para uso en todos los pacientes con LES; los estudios de observación
han demostrado que el tratamiento con hidroxicloroquina se asocia con una mayor
supervivencia, una menor actividad de la enfermedad, la prevención del daño
orgánico y la prevención de la trombosis. 10,11 La prevención de la trombosis puede
ocurrir debido a las reducciones resultantes en la activación plaquetaria y la
disfunción endotelial. 12
El tratamiento adicional para el LES depende de las
manifestaciones y la actividad específicas de la enfermedad. El diagnóstico de
endocarditis trombótica no bacteriana no justifica por sí mismo el tratamiento
inmunosupresor. Esta paciente fue evaluada por otras manifestaciones de órganos
diana, incluida la enfermedad renal, que no estaba presente. Debido a que no
hubo otros signos de compromiso orgánico activo, no fue tratado con
glucocorticoides sistémicos o un inmunosupresor ahorrador de glucocorticoides.
Sin embargo, requerirá un seguimiento a largo plazo para el desarrollo de otras
manifestaciones de lupus.
MANEJO DE NEUROLOGÍA
Los síntomas del paciente de debilidad facial en el
lado derecho y habla ininteligible se localizaron en la corteza frontal
posterior izquierda. La breve duración de los síntomas sugería un AIT como
mecanismo, probablemente debido a un émbolo, dada la presencia de múltiples
infartos crónicos dispersos observados en las imágenes de la cabeza. La
ausencia de lesiones isquémicas agudas en la RM potenciada por difusión no
descarta el AIT, ya que las lesiones isquémicas suelen desarrollarse después de
una isquemia más prolongada o grave.
En adultos jóvenes y de mediana edad, la isquemia
cerebral suele deberse a causas relativamente poco comunes de accidente
cerebrovascular, como las arteriopatías cerebrales no ateroscleróticas; tumores
cardíacos, endocarditis valvular o embolia paradójica a través de un agujero
oval permeable; y condiciones hematológicas, infecciosas, reumatológicas y
otras. 13 En este paciente, la amplia distribución temporal y espacial de las
lesiones isquémicas y la ausencia de anomalías arteriales en la Angio-TC de
cabeza y cuello sugerían una embolia cardíaca en curso. 14 De hecho, las
imágenes cardíacas revelaron vegetaciones valvulares que eran consistentes con
SAF asociado a endocarditis trombótica no bacteriana.
La paciente fue evaluada en el segundo hospital dentro
de 1 hora después del inicio de los síntomas del accidente cerebrovascular. Las
terapias para accidentes cerebrovasculares agudos, como el activador tisular
del plasminógeno intravenoso y la trombectomía intraarterial, son efectivas y
probablemente seguras en la población de adultos jóvenes y de mediana edad. Sin
embargo, estas terapias se suspendieron, ya que los déficits del paciente eran
leves y luego se resolvieron por completo y no hubo evidencia de un trombo en
la angio-TC. Inicialmente fue tratada con aspirina y atorvastatina para la
prevención secundaria del accidente cerebrovascular. Estudios recientes han
demostrado un beneficio con la terapia antiplaquetaria dual a corto plazo en
pacientes con ictus leve y AIT, 15pero este paciente pasó al tratamiento con
heparina intravenosa cuando se hizo evidente el diagnóstico de endocarditis
trombótica no bacteriana. Aunque las pautas basadas en la evidencia no
respaldan el uso de heparina en el accidente cerebrovascular agudo, creemos que
el beneficio supera el riesgo en ciertas afecciones, 16 incluida la
endocarditis trombótica no bacteriana, que conlleva un riesgo particularmente
alto de accidente cerebrovascular embólico recurrente.
Para la prevención a largo plazo del accidente
cerebrovascular en esta paciente, recomendamos la anticoagulación oral con un
antagonista de la vitamina K (en lugar de un anticoagulante oral directo) por
tiempo indefinido si el sangrado vaginal no se vuelve más problemático.
Desafortunadamente, los pacientes jóvenes y de mediana edad con accidente
cerebrovascular e incluso AIT se enfrentan a importantes desafíos psicosociales
y vocacionales de por vida, y se informa que la mortalidad en esta población
llega al 25% en un período de 20 años. 13 Por lo tanto, brindamos consejería y
seguimiento recomendado con un trabajador social. Finalmente, asesoramos a la
paciente sobre el reconocimiento del inicio del accidente cerebrovascular, la
eficacia dependiente del tiempo de las terapias para el accidente
cerebrovascular agudo y las formas de minimizar el riesgo de accidente
cerebrovascular (p. ej., ejercicio regular, evitar fumar y evitar la terapia
con estrógenos).
MANEJO DE LA ENDOCARDITIS TROMBÓTICA NO BACTERIANA
El primer paso en el tratamiento de la endocarditis
trombótica no bacteriana es identificar y tratar la causa subyacente. Faltan
datos aleatorios que respalden el uso de terapia antiplaquetaria o
anticoagulante en ausencia de embolia. Una guía basada en la evidencia
recomienda el uso de anticoagulación sistémica con heparina de bajo peso
molecular o heparina no fraccionada si hay evidencia de endocarditis trombótica
no bacteriana y embolismo pulmonar o sistémico. 17Sin embargo, sobre la base de
datos retrospectivos que muestran altas tasas de embolización, algunos expertos
recomiendan el uso de warfarina indefinidamente, independientemente de la
evidencia de embolia. Además, faltan ensayos prospectivos que evalúen las
indicaciones de la cirugía, así como su efectividad, por lo que la decisión de
realizarla es individualizada. La mayoría de los expertos recomendarán la
cirugía para indicaciones similares a la endocarditis infecciosa, como
insuficiencia cardíaca debida a disfunción valvular, perforación aguda de
valvas y prevención de embolización recurrente, con anticoagulación
posoperatoria continua con warfarina. 18
Establecer pautas para el seguimiento de los pacientes
es un desafío, porque no se ha demostrado que la presencia de enfermedad
valvular esté temporalmente relacionada con las características clínicas del
LES y la actividad de la enfermedad. 19 Aunque no existe una recomendación
formal con respecto a las imágenes ecocardiográficas de vigilancia en pacientes
en quienes se diagnostica LES, para aquellos en quienes se diagnostica
endocarditis trombótica no bacteriana, algunos expertos sugieren repetir las
imágenes cada 6 semanas a 3 meses para evaluar la progresión o resolución de
las vegetaciones y por la presencia de disfunción valvular. Los pronósticos
informados para pacientes con LES y endocarditis trombótica no bacteriana son
malos. En un estudio transversal longitudinal de 6 años, el 16 % de los
pacientes sufrieron un accidente cerebrovascular nuevo o recurrente, el 14 %
presentaron discapacidad cognitiva y el 9 % fallecieron. 20
MANEJO DE GINECOLOGÍA
Se consultó al equipo ginecológico para obtener ayuda
con el manejo del sangrado menstrual abundante y los fibromas en una paciente
que necesitaba anticoagulación a largo plazo en el contexto de LES, SAF y
accidentes cerebrovasculares embólicos relacionados con endocarditis trombótica
no bacteriana. Las imágenes de ultrasonido pélvico mostraron un útero
agrandado, que medía 11 cm por 10 cm por 8 cm, y dos fibromas (uno de 5,0 cm de
diámetro y el otro de 5,6 cm de diámetro). Los fibromas probablemente eran de
origen intramural, con un componente submucoso, y parecían haber distorsionado
el endometrio. Dados los síntomas de AIT de esta paciente, inicialmente se
suspendió el tratamiento con acetato de medroxiprogesterona. Afortunadamente,
su abundante sangrado se había resuelto espontáneamente, a pesar de que había
recibido heparina. Se realizó una evaluación ginecológica adicional;
Elegimos colocar un dispositivo intrauterino (DIU)
liberador de levonorgestrel, ya que se asocia con una baja absorción sistémica,
reduce la pérdida de sangre menstrual en pacientes que reciben anticoagulación,
21 y tiene un nivel de riesgo de categoría 2 (lo que indica que sus ventajas
superan sus riesgos) de los Centros para el Control y la Prevención de
Enfermedades de EE. UU. Criterios médicos de elegibilidad para la
anticoncepción (US MEC) en pacientes con trombosis venosa profunda aguda y
embolia pulmonar. En pacientes con LES en las que están presentes anticuerpos
antifosfolípidos, los métodos anticonceptivos de progestina sola y el DIU
liberador de levonorgestrel tienen cada uno un nivel de riesgo de categoría 3
de MEC de EE. UU. (lo que indica que los riesgos teóricos o comprobados de
estos anticonceptivos generalmente superan las ventajas), dada mayor riesgo de
tromboembolismo venoso.
Desafortunadamente, después de que la paciente fue
dada de alta, dos DIU liberadores de levonorgestrel fueron expulsados
espontáneamente durante dos episodios separados de sangrado abundante. Luego
eligió recibir acetato de medroxiprogesterona de depósito por vía intramuscular
1 mes y 3 meses después de la presentación actual, con una reducción posterior
de la menstruación. Dado su deseo de evitar la intervención quirúrgica con
histerectomía debido a preocupaciones sobre la menopausia y la necesidad de
extirpación de ovarios, se sometió a una embolización de fibroma uterino 7
meses después de la presentación inicial en este hospital. Con el tiempo, la
paciente ha optado por posponer la terapia hormonal adicional debido a los
molestos efectos secundarios. Ella tiene la esperanza de una reducción en el
sangrado después del procedimiento de embolización.
SEGUIMIENTO
Un examen de seguimiento 6 semanas después del alta de
este hospital reveló una nueva artritis inflamatoria de codos, rodillas y
tobillos, consistente con una manifestación clínica adicional de LES. La
paciente continuó el tratamiento con hidroxicloroquina, con una notable
disminución de las erupciones y de los síntomas del fenómeno de Raynaud. Un
ecocardiograma transtorácico de seguimiento no mostró un aumento en la
vegetación mitral y la regurgitación mitral se redujo a leve. Ocho meses después
de la presentación en este hospital, el paciente está relativamente bien y
continúa recibiendo terapia con hidroxicloroquina para el LES. Las pruebas
repetidas para anticuerpos SAF han permanecido como "triple
positivas". Ella también continúa recibiendo terapia de anticoagulación
con un plan para continuar indefinidamente.
DIAGNOSTICO FINAL
LUPUS ERITEMATOSO SISTÉMICO CON SÍNDROME
ANTIFOSFOLÍPIDO.
Traducción de:
A 41-Year-Old Woman with Transient Ischemic Attack and
Mitral Valve Masses
Mark A. Matza, M.D., Sandra P. Rincon, M.D., Evin
Yucel, M.D., April M. Jorge, M.D., Aneesh B. Singhal, M.D., Carrie A. Coleman,
M.D., and Sacha N. Uljon, M.D., Ph.D.
https://www.nejm.org/doi/full/10.1056/NEJMcpc2115855?query=featured_home
References
1. Bushnell C, Chireau M. Preeclampsia
and stroke: risks during and after preg[1]nancy.
Stroke Res Treat 2011;2011:858134.
2. Miyakis S, Lockshin MD, Atsumi T,
et al. International consensus statement
on an update of the classification criteria
for definite antiphospholipid syndrome
(APS). J Thromb Haemost 2006;4:295-306.
3. Libman E, Sacks B. A hitherto unde[1]scribed
form of valvular and mural endo[1]carditis.
Arch Intern Med 1924;33:701-37.
4. Roldan CA, Shively BK, Crawford MH.
An echocardiographic study of valvular
heart disease associated with systemic
lupus erythematosus. N Engl J Med 1996;
335:1424-30.
5. Elman SA, Joyce C, Braudis K, et al.
Creation and validation of classification
criteria for discoid lupus erythematosus.
JAMA Dermatol 2020;156:901-6.
6. Devreese KMJ, Ortel TL, Pengo V, de
Laat B; Subcommittee on Lupus Anti[1]coagulant/Antiphospholipid
Antibodies.
Laboratory criteria for antiphospholipid
syndrome: communication from the SSC
of the ISTH. J Thromb Haemost 2018;16:
809-13.
7. Naides SJ, Genzen JR, Abel G, Bashle[1]ben
C, Ansari MQ. Antinuclear antibodies
testing method variability: a survey of
participants in the College of American
Pathologists’ proficiency testing program.
J Rheumatol 2020;47:1768-73.
8. Egner W. The use of laboratory tests
in the diagnosis of SLE. J Clin Pathol
2000;53:424-32.
9. Insfrán CE, Aikawa NE, Pasoto SG,
et al. 2019-EULAR/ACR classification cri[1]teria
domains at diagnosis: predictive fac[1]tors
of long-term damage in systemic lu[1]pus
erythematosus. Clin Rheumatol 2021;
41:1079-85
MH, Venuturupalli SR. New insights into
mechanisms of therapeutic effects of anti[1]malarial
agents in SLE. Nat Rev Rheuma[1]tol
2012;8:522-33.
12. Cohen H, Cuadrado MJ, Erkan D,
et al. 16th International Congress on Anti[1]phospholipid
Antibodies Task Force re[1]port
on antiphospholipid syndrome treat[1]ment
trends. Lupus 2020;29:1571-93.
13. Singhal AB, Biller J, Elkind MS, et al.
Recognition and management of stroke
in young adults and adolescents. Neurol[1]ogy
2013;81:1089-97.
14. Ay H, Benner T, Arsava EM, et al. A
computerized algorithm for etiologic clas[1]sification
of ischemic stroke: the Caus[1]ative
Classification of Stroke System.
Stroke 2007;38:2979-84.
15. Kleindorfer DO, Towfighi A, Chaturvedi
S, et al. 2021 guideline for the prevention
of stroke in patients with stroke and tran[1]sient
ischemic attack: a guideline from
the American Heart Association/American
Stroke Association. Stroke 2021;52(7):e364-
e467.
16. Rocha EA, Ji R, Ay H, et al. Reduced
ischemic lesion growth with heparin in
acute ischemic stroke. J Stroke Cerebro[1]vasc
Dis 2019;28:1500-8.
17. Whitlock RP, Sun JC, Fremes SE, Ru[1]bens
FD, Teoh KH. Antithrombotic and
thrombolytic therapy for valvular disease:
antithrombotic therapy and prevention of
thrombosis, 9th ed: American College of
Chest Physicians evidence-based clinical
practice guidelines. Chest 2012;141:Suppl:
e576S-e600S.
18. Zmaili MA, Alzubi JM, Kocyigit D,
et al. A contemporary 20-year Cleveland
Clinic experience of nonbacterial throm[1]botic
endocarditis: etiology, echocardio[1]graphic
imaging, management, and out[1]comes.
Am J Med 2021;134:361-9.
19. Ong ML, Veerapen K, Chambers JB,
Lim MN, Manivasagar M, Wang F. Car[1]diac
abnormalities in systemic lupus ery[1]thematosus:
prevalence and relationship
to disease activity. Int J Cardiol 1992;34:
69-74.
20. Roldan CA, Sibbitt WL Jr, Qualls CR,
et al. Libman-Sacks endocarditis and em[1]bolic
cerebrovascular disease. JACC Car[1]diovasc
Imaging 2013;6:973-83.
21. Chi C, Huq FY, Kadir RA. Levonorges[1]trel-releasing
intrauterine system for the
management of heavy menstrual bleeding
in women with inherited bleeding disor[1]ders:
long-term follow-up. Contraception
2011;83:242-7






