En este ejercicio clínico se presenta un caso que es discutido por un médico internista al que se le van proporcionando datos de la historia clínica en forma secuencial, y este analiza el cuadro a la luz de los nuevos elementos, de una manera análoga al proceso diagnóstico en la práctica real de la medicina.




PRESENTACIÓN
Un hombre de 28 años que acude al servicio de
urgencias con dolor abdominal náuseas y vómitos. El paciente informó que había
tenido dolor intermitente en los
cuadrantes superior e inferior
derechos del abdomen durante 6 semanas. El dolor empeoraba cuando comía
alimentos y generalmente disminuía en 1 a 2 horas después de tomar antiácidos.
En esta ocasión, el dolor había sido severo y había durado 8 horas. La ingesta
de alimentos empeoró el dolor y el cual fue seguido por un episodio de vómitos no
hemáticos ni biliosos. No tenía fiebre o escalofríos.
PONENTE
Los episodios recurrentes y limitados de dolor en el
lado derecho del abdomen después de las comidas son característicos del cólico
biliar y de la enfermedad de ulcerosa péptica. El aumento de la severidad y la
persistencia del dolor en el momento de la presentación actual del paciente
podría representar evolución a colecistitis aguda o a perforación ulcerosa.
Otras causas de dolor en el lado derecho del abdomen incluyen apendicitis, hepatitis, colitis,
nefrolitiasis, e infarto renal, aunque estos trastornos parecen ser poco
probables sobre la base de la naturaleza
recurrente y previamente limitada del dolor.
EVOLUCIÓN
El paciente había recibido un diagnóstico de
síndrome de Gilbert 2 años antes cuando su médico de atención primaria notó que
tenía hiperbilirrubinemia indirecta aislada. Él paciente era obeso (índice de masa corporal de 33) y
tenía hígado graso no alcohólico. Nunca
había usado tabaco o drogas recreativas y rara vez bebía alcohol. En el examen
físico, la temperatura fue de 37.1 ° C, la frecuencia cardíaca de 84 latidos
por minuto, la presión arterial 145/90 mm Hg, la frecuencia respiratoria 18
respiraciones por minuto, y la saturación de oxígeno de 100% mientras respiraba
aire ambiente. Se sentía incómodo por el dolor abdominal. Había ictericia en
escleras. Los exámenes cardíaco y pulmonar fueron normales. Había dolor leve
en los cuadrantes superior e inferior derechos del abdomen, sin rebote ni reacción peritoneal. No había estigmas de
enfermedad hepática crónica.
PONENTE
Las causas más comunes de hiperbilirrubinemia
indirecta aislada en un adulto son el síndrome de Gilbert y la hemólisis. El hígado
graso no alcohólico puede estar asociado con una incomodidad sorda en el cuadrante
superior derecho, pero no suele ser causa de dolor intenso. La obesidad y la hemólisis
predisponen a los pacientes a la formación de cálculos biliares (de colesterol
y pigmentarios respectivamente). La
ictericia del paciente y el dolor en el lado derecho del abdomen sugieren un
trastorno hepatobiliar como hepatitis, infiltración hepática, congestión
hepática, colecistitis o coledocolitiasis.
EVOLUCIÓN
El recuento de leucocitos fue de 19.800 por
milímetro cúbico (86% de neutrófilos, 5% de linfocitos, 8% de monocitos y cero
bandas), el nivel de hemoglobina de 16.0 g por decilitro, y el recuento de
plaquetas es de 196,000 por milímetro cúbico. Los niveles de electrolitos,
nitrógeno ureico en sangre, creatinina y glucosa eran normales. El total el
nivel de bilirrubina fue de 6,7 mg por decilitro por litro; rango de referencia, 0.2 a 1.1 mg
por decilitro), y la bilirrubina directa
era de 2,6 mg por decilitro (rango de referencia, de 0,0 a
0,3 mg por decilitro). La alanina aminotransferasa fue de 185 U por litro
(valor normal, menos de 54), la aspartato aminotransferasa 157 U por litro (rango
normal, 10 a 42) y la fosfatasa alcalina 101 U por litro (rango normal, 40 a
130). La albúmina era de 4.7 g por decilitro. El RIN fue 1.1. La lipasa era
normal. La tomografía computarizada de abdomen reveló un hígado de tamaño
normal con esteatosis leve y esplenomegalia (16 cm) en el diámetro sagital)
pero no había cálculos biliares, ni dilatación de la vía biliar ni evidencia de
colecistitis. El apéndice no fue
visualizado. El paciente fue ingresado en el hospital y se comenzó con la administración
intravenosa de meropenem.
PONENTE
La hiperbilirrubinemia predominantemente indirecta sugiere
que el síndrome de Gilbert o la hemólisis
pueden estar acompañando a una
enfermedad aguda. Sin embargo, un dolor en el lado derecho del abdomen,
leucocitosis, y un nivel elevado de bilirrubina directa (proporcional a los
otros valores bioquímicos del hígado) despiertan preocupación por colecistitis o coledocolitiasis. Los resultados de la TC no sugieren ninguna condición
categóricamente, pero la ultrasonografía es más sensible para la detección de
cálculos biliares dentro de la vesícula biliar, y la colangiografía por
resonancia magnética para detectar o descartar coledocolitiasis de forma más definitiva. Aunque
el estrés fisiológico y la deshidratación puede contribuir a la leucocitosis,
descartar la colangitis es esencial. La hemólisis a veces causa una modesta
elevación de los niveles de aminotransferasas (particularmente de aspartato
aminotransferasa) además de hiperbilirrubinemia indirecta. La hemoglobina
normal no es compatible con un diagnóstico de hemólisis, pero el paciente puede
tener hemoconcentración o hemólisis totalmente compensada. Deberían solicitarse un frotis de sangre
periférica y un recuento de
reticulocitos.
EVOLUCIÓN
En el segundo día de hospital, el dolor abdominal disminuido
y el recuento de glóbulos blancos disminuyó a 14.200 por milímetro cúbico, pero
los valores del hepatograma aumentaron. La bilirrubina total fue de 19,5 mg por decilitro
y el nivel de bilirrubina directa de 11,2 mg por decilitro. La alanina
aminotransferasa era de 519 U por
litro, aspartato aminotransferasa nivel
188 U por litro, y la fosfatasa alcalina de 171 U por litro. La prueba de
anticuerpos de superficie de hepatitis B fue positivo. Antígeno de superficie
de Hepatitis B y anticuerpos para hepatitis A (IgM), hepatitis C y antígeno core
de hepatitis B fueron negativos. La
ceruloplasmina era normal. Los FAN y los anticuerpos antimitocondriales fueron
negativos. La ultrasonografía de abdomen reveló una vesícula llena de barro
biliar con engrosamiento de la pared y líquido pericolecístico, hallazgos consistente
con colecistitis aguda (Fig. 1A). No había dilatación de la vía biliar común
del conducto ni coledocolitiasis. La colangiopancreatografía por resonancia magnética confirmó estos
hallazgos (Fig. 1B). Infiltración grasa del hígado y esplenomegalia se observaron
en ambos estudios. Las venas porta y hepáticas eran permeables y su flujo era
normal en la ecografía.
Figura 1.
Ultrasonografía y Colangiopancreatografía de la vesícula
biliar.
Una imagen ultrasonográfica del abdomen (Panel A) muestra
una vesícula biliar llena de barro biliar (flecha), con engrosamiento de la
pared de la vesícula biliar (asteriscos) y una pequeña cantidad de líquido
pericolecístico (puntas de flecha). En la imagen de RMN en T2 con supresión
grasa (Panel B) muestra engrosamiento de la pared de la vesícula biliar e
irregularidad con líquido pericolecístico asociado (puntas de flecha) y una
cantidad moderada de barro en la vesícula biliar (flecha).
PONENTE
El aumento brusco en el nivel de bilirrubina directa
puede ser explicado por la colecistitis aguda con gran inflamación (síndrome de
Mirizzi), y/o transitoria obstrucción de
la vía biliar por barro biliar o microlitiasis, superpuesta a la hepatopatía
crónica del paciente ( hígado graso no alcohólico). La ultrasonografía y la
colangioresonancia magnética mostraron colecistitis aguda que puede causar ictericia leve incluso en
ausencia de obstrucción de la vía biliar principal. La coledocolitiasis causa
ictericia más franca aunque en estos casos el nivel de la bilirrubina directa es
típicamente inferior a 10 mg por decilitro. Es posible que tanto el barro
biliar como un cálculo biliar
transitoriamente obstruyan la vía biliar
biliar común antes de que el método más
sensible de imágenes detecte la causa por ejemplo coledocolitiasis. Estos
estudios diagnósticos se realizaron en
el segundo día de hospital; la reducción del dolor apoya esa hipótesis. El
nivel de bilirrubina indirecta permanece alto, pero la hemólisis o el síndrome
de Gilbert rara vez elevan la
bilirrubina indirecta a más de 5 mg por
decilitro. La combinación de enfermedad hepática y hemólisis en un paciente
joven debe impulsar la consideración de enfermedad de Wilson. Sin embargo, el
paciente no tiene un desproporcionado aumento de la aspartato aminotransferasa
ni de la alanina aminotransferasa que son típicos de la enfermedad de Wilson, y
por otro lado el nivel de ceruloplasmina es normal. Dado el elevado nivel de
aminotransferasas, es importante considerar causas virales, tóxicas, vasculares
y autoinmunes de hepatitis. El panel estándar para hepatitis virales fue negativo, aunque falsos
negativos pueden ocurrir. La prueba de reacción en cadena de la polimerasa
pueden complementar al panel básico utilizado para hepatitis cuando se sospecha fuertemente
infección viral (por ej infección aguda por el virus de la hepatitis C).
Pruebas para virus que ocasionalmente infectan el hígado como citomegalovirus,
virus de Epstein-Barr y virus del herpes simple: también deben tenerse en
cuenta. Sería útil preguntar al paciente sobre el uso de acetaminofeno, otros medicamentos
de venta libre y suplementos nutricionales que podría causar efectos
hepatotóxicos. No existían antecedentes de hipotensión arterial severa u
obstrucción de la vena hepática observados en la ecografía que pudiesen sugerir una causa vascular de la lesión
hepática.
La esplenomegalia es poco probable que sea
atribuible a la hipertensión portal, dado el nivel normal de albúmina RIN normal, y recuento de
plaquetas normal. proporción de albúmina, relación internacional normalizada.
La esplenomegalia podría sugerir en cambio infiltración (por ejemplo, infección
o cáncer) o acumulación de glóbulos rojos anormales en el contexto de
enfermedad hemolítica
EVOLUCIÓN
En el tercer día de hospital, el dolor abdominal se
resolvió, y el paciente permaneció afebril. Los niveles de bilirrubina total
bajaron a 5.7 mg por decilitro y el
nivel de bilirrubina directa a 2,5 mg por decilitro. La alanino aminotransfersa
alanine fue de 292 U por litro, el nivel
de aspartato aminotransferasa 89 U por litro, y el nivel de fosfatasa alcalina
174 U por litro. Meropenem se suspendió. El paciente fue dado de alta a su casa,
y remitido a cirugía para una colecistectomía electiva, y programada y con una
cita para medicina general al día siguiente.
PONENTE
El cuadro clínico es consistente con colecistitis
aguda y obstrucción biliar transitoria superpuesta (por ejemplo, por pasaje de
barro o cálculos biliares). La resolución rápida es más típica del barro biliar
que el de una hepatitis viral o tóxica
EVOLUCIÓN
En la consulta clínica del día siguiente, un médico que no
había conocido al paciente durante la internación, revisó los resultados de sus
prueba de 2 años antes. En dichas pruebas, junto a una bilirrubina indirecta de
1,9 mg por decilitro, el paciente había tenido una lactato deshidrogenasa
elevada en un nivel de 320 U por litro (rango normal, 120 a 220) y un nivel de
haptoglobina de menos de 8 mg por decilitro (rango normal, 26 a 240). Sus
niveles de aspartato aminotransferasa, alanina aminotransferasa, y la fosfatasa
alcalina eran normales en ese momento. El nivel de hemoglobina era de 15.0 g
por decilitro, y el volumen corpuscular medio fue de 83.6 fl, con una
concentración media de hemoglobina corpuscular de 37 g por decilitro (rango
normal, 32 a 36) y un ancho de distribución de glóbulos rojos del 15.7% (normal
rango, 11.5 a 14.5). La revisión del informe de una ecografía de 2 años antes reveló hepatomegalia
con infiltración de hígado graso y esplenomegalia (16 cm en el diámetro
sagital). No había cálculos biliares.
PONENTE
La hiperbilirrubinemia indirecta sostenida, una
elevación del nivel de lactato deshidrogenasa, un indetectable nivel de
haptoglobina y esplenomegalia con una hemoglobina normal sugiere una hemólisis compensada. La
anemia hemolítica puede ser causada por procesos inmunes, microangiopáticos o
infecciosos, o puede ser el resultado de anormalidades congénitas de la
membrana de los eritrocitos, de las enzimas o de la hemoglobina tales como son
la esferocitosis hereditaria, la deficiencia de glucosa 6-fosfato
deshidrogenasa, la anemia drepanocítica y la talasemia. La elevada concentración de
hemoglobina corpuscular media es particularmente característica de la
esferocitosis hereditaria
EVOLUCIÓN
Estudios de laboratorio de la visita clínica después
del alta revelaron un porcentaje de reticulocitos del 3.2% (rango normal, 0.5 a
2.2) y un recuento absolutos de reticulocitos de 172.100 por milímetro cúbico (normal rango,
31,500 a 108,800). El frotis de sangre periférica obtenido en esa cita y en el primer
día de hospital fueron revisados por un hematopatólogo y mostró los cuerpos de
Pappenheimer (gránulos que contienen hierro) pero no había acantocitos, esquistocitos
o esferocitos. En esa misma cita, la madre del paciente recordó que el padre
del paciente había sido sometido a una colecistectomía por cálculos biliares a
los 27 años de edad.
PONENTE
La reticulocitosis sostenida en este paciente es una
respuesta compensatoria a la hemólisis en curso. El antecedente de cálculos biliares de su padre a una edad
temprana sugiere una anemia hemolítica hereditaria con cálculos biliares
pigmentarios. Anemia falciforme, talasemias, y esferocitosis hereditaria causa colelitiasis
prematura, pero el frotis de sangre periférica no muestra evidencia de estas
condiciones. Además, las talasemias se caracterizan por microcitosis y la talasemia
sintomática y la enfermedad de células falciformes típicamente se manifiesta en
la infancia y están asociados con anemia. El paciente probablemente tuvo una
lesión hepática transitoria de obstrucción de la vía biliar por barro biliar o
un cálculo secundario a un estado hemolítico hereditario crónico. Lo atípico de
la obstrucción biliar transitoria en este paciente es la ausencia de dilatación
de la vía biliar común en múltiples estudios de imágenes, e hiperbilirrubinemia
marcada, aunque esta última probablemente sea multifactorial. La historia familiar, la concentración hemoglobínica
corpuscular media elevada y la hemólisis
crónica compensada con una predisposición hacia la formación de cálculos
biliares apuntan a un diagnóstico de esferocitosis hereditaria. Los esferocitos
son casi universales en la esferocitosis hereditaria pero pueden no ser vistos en
el frotis de sangre periférica en casos leves en
los que el número de glóbulos rojos deformados es bajo.
EVOLUCIÓN
Cuatro semanas después, el paciente se sometió a una
colecistectomía laparoscópica, que
reveló múltiples cálculos biliares pigmentarios, el más grande de 1,0 cm por 0,6 cm por 0,6 cm. Un
frotis de sangre periférica perioperatorio que fue revisado por un segundo hematopatólogo
mostró esferocitos ocasionales. Un mes después de la colecistectomía, el
paciente vio a un hematólogo. Los valores de laboratorio de esa visita
mostraron una normalización del nivel de aminotransferasas y un retorno a los valores basales de bilirrubina total (3.3 mg por decilitro)
con una bilirrubina directa de 0,9 mg por
decilitro. La prueba de fragilidad
osmótica reveló una mayor fragilidad de los eritrocitos, y la determinación por
citométría de flujo de colorante eosina-5'-maleimida (EMA) mostraron una
disminución de la fluorescencia, que
confirmó el diagnóstico de esferocitosis hereditaria. La prueba de
antiglobulina directa (reacción de Coombs), no se realizó. Un frotis de sangre
periférica que se obtuvo 7 meses después mostró múltiples esferocitos (Fig. 2).

Figura 2. Frotis de sangre periférica.
Un frotis de sangre periférica (tinción de Wright)
muestra esferocitos (selectivamente indicado por flechas).
Para evaluar el síndrome de Gilbert concomitante (uridine
diphosphoglucuronato-glucuronosyltransferase), se realizó el genotipado 1A1
(UGT1A1). El resultado mostró heterocigosidad para el polimorfismo TA7. Aunque
los niveles de bilirrubina pueden estar ligeramente elevados en estados heterocigotos, se requiere
típicamente homocigosidad para
diagnosticar síndrome de Gilbert definitivamente. El aumento crónico de
la bilirrubina indirecta en este paciente fue atribuido a
esferocitosis hereditaria.
Siete meses después, el paciente se sentía bien, sin recurrencia de su dolor abdominal
COMENTARIO
Una exacerbación aguda de un cuadro de dolor
intermitente en el lado derecho del abdomen asociado con hiperbilirrubinemia
y aminotransferasas elevadas sugirieron un evento común por su prevalencia:
colecistitis aguda con obstrucción biliar transitoria por barro biliar o
cálculos. Sin embargo, habías algunas pistas ocultas o latentes en su historia
clínica, que incluía el laboratorio donde existía evidencia de hemólisis, una concentración
de hemoglobina corpuscular media alta y
una esplenomegalia que mirados en conjunto sugirieron una causa poco común del cuadro: la esferocitosis
hereditaria.
La esferocitosis hereditaria es la anemia hemolítica
causada por un defectos en las proteínas de membranas de glóbulos rojos más
común, con una incidencia de 1 caso por 2000 personas de ascendencia del norte
de Europa.1 En la esferocitosis hereditaria, deficiencias en las proteínas de membrana
o en las proteínas del citoesqueleto proteínas
citoesqueléticas asociadas, secundarias a mutaciones genéticas heredadas dan como
resultado una interacción debilitada entre la membrana y el citoesqueleto (Fig. 3).
Esto lleva a la degradación de la membrana mediante vesiculación y pérdida
progresiva de la membrana, a medida que los glóbulos rojos pasan a través de
los senos esplénicos. La forma esférica resultante conduce a una menor
deformabilidad y al aspecto característico en el frotis de sangre periférica. Estos esferocitos tienen dificultad para pasar a
través de las estrechas fenestraciones de los sinusoides venosos y son
destruidos por los macrófagos dentro del bazo.1
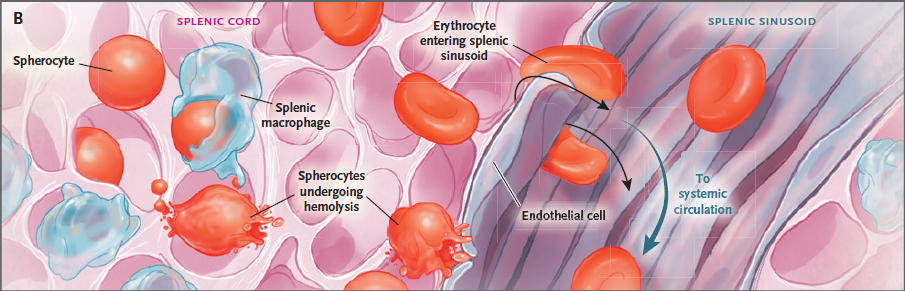
Figura 3. Hemólisis en la esferocitosis hereditaria.
El panel A muestra la estructura de un glóbulo rojo
normal y un esferocito; los insertos muestran interacciones entre la membrana y
el citoesqueleto mediados por las proteínas de banda 3, banda Rh, y banda 4.(2),
y anquirina; la glicoforina C, banda 3,
Rh, banda 4 (3), y actina; α-espectrina y β-espectrina.
Las mutaciones genéticas de cualquiera de las
proteínas sombreadas pueden conducir a deficiencias que causan esferocitosis
hereditaria. La interrupción de la conexión entre la bicapa lipídica y el
citoesqueleto produce microvesiculación, pérdida de membrana y formación de
esferocitos.
El panel B muestra
la pulpa roja del bazo en la que los glóbulos rojos pasan de los senos a
los sinusoides a través de espacios estrechos entre las células endoteliales.
Los esferocitos son menos deformables debido a una reducida relación
superficie/volumen retiénendose
selectivamente en los senos , donde se produce destrucción adicional o
hemólisis.
Los pacientes con esferocitosis hereditaria
típicamente se presentan con anemia
hemolítica, ictericia, esplenomegalia, una concentración de hemoglobina
corpuscular media elevada y
esferocitosis. La esferocitosis hereditaria leve, que este paciente tenía, se
distingue de la forma moderada y severa de esferocitosis hereditaria por el
grado de anemia (nivel de hemoglobina, por lo general de 11 a 15 g por
decilitro), reticulocitosis (porcentaje de reticulocitos, generalmente 3% a
6%), bilirrubinemia generalmente de 1 a 2 mg por decilitro y esferocitosis
(generalmente de 2 a 3 esferocitos) por campo de gran aumento) .1
Tradicionalmente, la medición de la hemólisis en una solución hipotónica (la
prueba de fragilidad osmótica), se ha utilizado para confirmar el diagnóstico
en pacientes en quienes las características estándar de laboratorio son
insuficientes. Sin embargo, los resultados de las pruebas son normales en 10 a
20% de los pacientes con esferocitosis hereditaria y pueden ser anormales en
pacientes con otras causas de esferocitosis, como la anemia hemolítica
autoinmune.2 La prueba de unión de EMA es un ensayo de citometría de flujo que detecta disminución de
la unión del colorante EMA a la proteína de la banda 3; la deficiencia de esta
proteína es una característica fisiopatológica central en la esferocitosis
hereditaria. El test de unión a EMA tiene una sensibilidad del 90% y una especificidad
del 95%, y es ahora el métrodo diagnóstico preferido en casos equívocos.3 La
esplenectomía es el tratamiento para los síntomas de la esferocitosis
hereditaria moderada o grave. 2 La esplenectomía no cambia las alteraciones del citoesqueleto de los eritrocitos, pero
elimina el foco primario de la hemólisis. El consenso actual desalienta la esplenectomía
en la esferocitosis hereditaria leve porque
los riesgos asociados con el inmunocompromiso resultante supera el riesgo de
complicaciones hemolíticas.2 En pacientes con esferocitosis hereditaria
moderada o grave, esta relación riesgo-beneficio se invierte porque la
esplenectomía disminuye sustancialmente
la hemólisis y la incidencia de cálculos biliares pigmentarios. La incidencia de
colelitiasis entre los niños y adultos jóvenes con esferocitosis hereditaria es
aproximadamente 40% .4, 5 La colecistectomía está generalmente reservada para pacientes con
síntomas de enfermedad colelitiásica. Estudios
retrospectivos han demostrado que los niños con esferocitosis hereditaria leve que
se someten a colecistectomía dirigida por síntomas, con poca frecuencia requieren esplenectomía posterior 6,7; estos
hallazgos sugieren que la esplenectomía en el momento de la colecistectomía no
está indicada en estos pacientes. Varias
oportunidades se perdieron en este paciente para establecer el
diagnóstico de esferocitosis hereditaria. El primer médico ambulatorio que lo vio dos años antes, probablemente consideró la hemólisis sobre la base de las pruebas que le solicitó, lactato deshidrogenasa y haptoglobina. Sin
embargo, el nivel normal de hemoglobina puede haber proporcionado una falsa confianza
del profesional y lo desalentaron en continuar por elcamino de la hemólisis como mecanismo de base y se conformó estableciendo un
diagnóstico del síndrome de Gilbert (aunque la esplenomegalia no podría ser
explicada por este diagnóstico). Los médicos que lo vieron durante la hospitalización no buscaron un diagnóstico unificador raro porque la obesidad del paciente proporcionó
un explicación de sus cálculos biliares. Finalmente, cuando la hemólisis fue
reconocida después del alta a través una
revisión retrospectiva de todos los datos tanto como paciente ambulatorio que
como internado, el hallazgo de los esferocitos que estaban inicialmente ausentes. Sin el conocimiento altamente
especializado de que los esferocitos pueden no ser visibles en tanto como el 2 a 3% de los pacientes con esferocitosis
hereditaria leve, era difícil de
defender este diagnóstico.8
Un
diagnóstico de esferocitosis hereditaria leve 2 años antes no habría
justificado una esplenectomía, ni colecistectomía, ni medicamentos o metodología de detección de
colelitiasis. Sin embargo, si el
paciente hubiese conocido su condición de esferocitosis hereditaria y así su riesgo asociado a padecer enfermedad de cálculos biliares, él podría
haberse presentado al sistema de
atención médica antes por su cuadro de 6 semanas de dolor abdominal episódico. La
ultrasonografía ambulatoria y una aceleración de la colecistectomía podrían haber evitado el costo y las complicaciones
asociadas con la colecistitis aguda y obstrucción transitoria del conducto
biliar común, que implicaban hospitalización, exposición a la radiación, riesgo
de pancreatitis y una incomodidad sustancial.
En retrospectiva, es fácil ver cómo el escrutinio de
la concentración de hemoglobina corpuscular media elevada o una investigación temprana sobre la
historia familiar podría haber apuntado a la esferocitosis hereditaria. Sin
embargo, los desafíos de reconocer la hemólisis sin anemia y reconocer
esferocitosis hereditaria sin esferocitos fueron importantes.
Todos los clínicos aspiran a discernir patrones de
diagnóstico cuando ellos se visibilizan por
primera vez, pero a veces el
derrotero hacia la verdad no es directo, sino que tenemos que ir y volver sobre
nuestros propios pasos, muchas veces girando en círculos muy redondos,
como esferocitos…
CONCLUSIONES DEL CASO
Otra vez un caso nos enseña cómo la mirada
retrospectiva que hurga en los antecedentes, en historias o registros pasados,
en causas de internaciones anteriores, o como en este caso, un dato de
bilirrubina indirecta ligeramente elevada en un laboratorio de dos años atrás, erróneamente
diagnosticado de síndrome de Gilbert. El médico que lo asistió en aquella
oportunidad estuvo muy cerca de arribar a un diagnóstico de anemia hemolítica
sobre la base de esplenomegalia, bilirrubina indirecta y LDH elevadas asociados
a haptoglobina baja, todos estudios solicitados apropiadamente y dirigidos a
confirmar las sospechas clínicas. Sin embargo, la ausencia de anemia y la inespecificidad
de los hallazgos del frotis de sangre periférica (la falta de microcitos,
hipocromía y target cells sugestivos de talasemia, la falta de drepanocitos
sugestivos de anemia de células falciformes, la ausencia de esferocitos
sugestivos de esferocitosis hereditaria o secundaria, la falta de esquistocitos
y plaquetopenia sugestivos de microangiopatía trombótica etc), desalentaron al
médico en su búsqueda, quien encontró en el síndrome de Gilbert una forma de
explicar la situación y tranquilizar su conciencia, al menos en parte ya que la
esplenomegalia ya presente en aquel momento quedaba sin aclarar. A veces es
necesario releer la historia pasada del paciente en lugar de solicitar nuevos
estudios para entender el caso desde una perspectiva más completa. Finalmente
digamos que este caso nos deja dos enseñanzas que es muy importante no olvidar,
en primer lugar que la ausencia de anemia no descarta hemólisis, la hemólisis
compensada es una situación perfectamente posible cuando la médula ósea alcanza
a reponer con su expansión eritroide el generalmente bajo nivel de hemólisis
como sucedió en este caso de esferocitosis leve. En segundo lugar que no se
puede descartar esferocitosis hereditaria sobre la base sólo de un frotis de
sangre periférica.Existe la posibilidad de esferocitosis sin esferocitos al menos
en algún punto de la evolución de la enfermedad.
Fuente:
CIRCLING BACK FOR THE DIAGNOSIS
Joseph Rencic, M.D., Mengyu Zhou, M.D., Gerald Hsu, M.D., Ph.D.,
and Gurpreet Dhaliwal, M.D.
References
1. Perrotta S,
Gallagher PG, Mohandas N.
Hereditary
spherocytosis. Lancet 2008;
372: 1411-26.
2. Bolton-Maggs
PH, Langer JC, Iolascon
A, Tittensor P,
King MJ. Guidelines
for the
diagnosis and management of
hereditary
spherocytosis — 2011 update.
Br J Haematol
2012; 156: 37-49.
3. Bianchi P, Fermo E, Vercellati C, et al.
Diagnostic power
of laboratory tests for
hereditary
spherocytosis: a comparison
study in 150
patients grouped according
to molecular and
clinical characteristics.
Haematologica
2012; 97: 516-23.
4. Bates GC, Brown
CH. Incidence of
gallbladder
disease in chronic hemolytic
anemia
(spherocytosis). Gastroenterology
1952; 21: 104-9.
5. Tamary H, Aviner S, Freud E, et al.
High incidence
of early cholelithiasis detected
by
ultrasonography in children and
young adults
with hereditary spherocytosis.
J Pediatr
Hematol Oncol 2003; 25: 952-4.
6. Ruparel RK,
Bogert JN, Moir CR, et al.
Synchronous
splenectomy during cholecystectomy
for hereditary
spherocytosis:
is it really
necessary? J Pediatr Surg 2014;
49: 433-5.
7. Alizai NK,
Richards EM, Stringer MD.
Is
cholecystectomy really an indication for
concomitant
splenectomy in mild hereditary
spherocytosis?
Arch Dis Child 2010;
95: 596-9.
8. Mariani M,
Barcellini W, Vercellati C,
et al. Clinical
and hematologic features of
300 patients
affected by hereditary spherocytosis
grouped
according to the type of
the membrane
protein defect. Haematologica
2008; 93:
1310-7.
4 comentarios:
Excelente artículo.
Muy bueno.
muy buen artículo sobre anatomía
Muy buen artículo, excelente información
Publicar un comentario